As
filed with the Securities and Exchange Commission on August 14, 2024
Registration
No. 333-
UNITED
STATES
SECURITIES
AND EXCHANGE COMMISSION
Washington,
D.C. 20549
FORM
S-1
REGISTRATION
STATEMENT UNDER THE SECURITIES ACT OF 1933
Tenon
Medical, Inc.
(Exact
name of registrant as specified in its charter)
| Delaware | | 3841 | | 45-5574718 |
(State or Other Jurisdiction of Incorporation or Organization) | | (Primary Standard Industrial Classification Code Number) | | (I.R.S. Employer Identification No.) |
104
Cooper Court
Los
Gatos, CA 95032
(408)
649-5760
(Address,
including zip code, and telephone number, including area code,
of
registrant’s principal executive offices)
Steven
M. Foster
Chief
Executive Officer and President
Tenon
Medical, Inc.
104
Cooper Court
Los
Gatos, CA 95032
(408)
649-5760
(Name,
address, including zip code, and telephone number, including area code, of agent for service)
Copies
to:
| Ross D. Carmel,
Esq. |
|
| Jeffrey P. Wofford, Esq. |
|
| Sichenzia Ross Ference
Carmel LLP |
|
| 1185 Avenue of Americas,
31st Floor |
|
| New York, New York 10036 |
|
| Telephone: (212) 930-9700 |
|
Approximate
date of commencement of proposed sale to the public: As soon as practicable after the effective date of this Registration Statement.
If
any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the
Securities Act of 1933 check the following box. ☒
If
this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the
following box and list the Securities Act registration statement number of the earlier effective registration statement for the same
offering. ☐
If
this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the
Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If
this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the
Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
Indicate
by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting
company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company”
in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ☐ | Accelerated filer | ☐ |
| Non-accelerated filer | ☒ | Smaller reporting company | ☒ |
| | Emerging growth company | ☒ |
If an emerging
growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any
new or revised financial accounting standards provided to Section 7(a)(2)(B) of the Securities Act. ¨
The
Registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the
Registrant shall file a further amendment which specifically states that this Registration Statement shall thereafter become effective
in accordance with Section 8(a) of the Securities Act of 1933 or until the Registration Statement shall become effective on such date
as the Commission acting pursuant to said Section 8(a), may determine.
The
information in this prospectus is not complete and may be changed. These securities may not be sold until the registration statement
filed with the Securities and Exchange Commission is effective. This prospectus is not an offer to sell these securities and is not soliciting
an offer to buy these securities in any state or other jurisdiction where the offer or sale is not permitted.
| PRELIMINARY
PROSPECTUS |
SUBJECT TO
COMPLETION |
DATED AUGUST
14, 2024 |
Up
to [*] shares of Common Stock
Common
Warrants to Purchase up to [*] shares of Common Stock
Pre-funded
Warrants to Purchase up to [*] shares of Common Stock
Up
to [*] shares of Common Stock underlying the Common Warrants
Up
to [*] shares of Common Stock underlying the Pre-funded Warrants

Tenon
Medical, Inc.
We
are offering on a best efforts basis up to [*] shares of our common stock, par value $0.001 per share, together with common warrants
to purchase up to [*] shares of common stock, (the “Common Warrants”), at an assumed combined public offering price of $[*]
per share and accompanying Common Warrant, based on the closing price of our common stock on The Nasdaq Capital Market, or Nasdaq, on
[*], 2024. Common Warrants are immediately exercisable on the date of issuance at an exercise price of $[*] per share (100% of the offering
price per share and accompanying Common Warrant) and will expire five years from the date of issuance. The shares of common stock and
Common Warrants are immediately separable and will be issued separately in this offering but must be purchased together in this offering.
We
are also offering pre-funded warrants (the “Pre-funded Warrants”) to purchase up to [*] shares of common stock to those purchasers
whose purchase of shares of common stock in this offering would result in the purchaser, together with its affiliates and certain related
parties, beneficially owning more than 4.99% (or, at the election of the purchaser, 9.99%) of our outstanding common stock immediately
following the consummation of this offering, in lieu of shares of common stock that would result in beneficial ownership in excess of
4.99% (or, at the election of the purchaser, 9.99%) of our outstanding common stock. Each Pre-funded Warrant will be immediately exercisable,
is exercisable for one share of our common stock and has an exercise price of $0.0001 per share. The Pre-Funded Warrants will expire
when exercised in full. Each Pre-funded Warrant is being offered together with the Common Warrants. The Pre-funded Warrants and Common
Warrants are immediately separable and will be issued separately in this offering but must be purchased together in this offering. For
each Pre-funded Warrant that we sell, the number of shares of common stock we are offering will be reduced on a one-for-one basis.
Pursuant
to the registration statement related to this prospectus, we are also registering the shares of common stock issuable upon exercise of
the Common Warrants and the Pre-Funded Warrants.
Our
common stock is listed on The Nasdaq Capital Market under the symbol “TNON.” The last reported sale price of our common stock
on The Nasdaq Capital Market on August, 2024 was $[*] per share.
We
do not intend to apply to list the Common Warrants or Pre-Funded Warrants on any national securities exchange or other nationally recognized
trading system. Without an active trading market, the liquidity of the Common Warrants and Pre-Funded Warrants will be limited.
The
public offering price for the securities in this offering will be determined at the time of pricing, and may be at a discount to the
current market price at the time. Therefore, the assumed public offering price used throughout this prospectus may not be indicative
of the final offering price. The final public offering price will be determined through negotiation between us, the placement agent and
the investors based upon a number of factors, including our history and our prospects, the industry in which we operate, our past and
present operating results, the previous experience of our executive officers and the general condition of the securities markets at the
time of this offering.
The
securities will be offered at a fixed price and are expected to be issued in a single closing. We expect this offering to be completed
not later than [one][two] business day[s] following the commencement of sales in this offering (the effective date of the registration
statement of which this prospectus forms a part) and we will deliver all securities to be issued in connection with this offering delivery
versus payment/receipt versus payment upon receipt of investor funds received by us. Accordingly, neither we nor the placement agent
have made any arrangements to place investor funds in an escrow account or trust account since the placement agent will not receive investor
funds in connection with the sale of the securities offered hereunder.
We
have engaged [*] (whom we refer to herein as “[*]” or the “Placement Agent”) as our exclusive placement agent
to use its reasonable best efforts to solicit offers to purchase our securities in this offering. The placement agent has no obligation
to purchase any of the securities from us or to arrange for the purchase or sale of any specific number or dollar amount of the securities.
Because there is no minimum offering amount required as a condition to closing in this offering, the actual public offering amount, placement
agent’s fee and proceeds to us, if any, are not presently determinable and may be substantially less than the total maximum offering
amounts set forth above and throughout this prospectus. We have agreed to pay the placement agent the placement agent fees set forth
in the table below. See “Plan of Distribution” in this prospectus for more information.
We
intend to use the proceeds from this offering for sales and marketing activities, including building a sales and marketing infrastructure,
training clinicians to use our products, clinical studies, and general corporate purposes, including working capital. See “Use
of Proceeds.”
Investing
in our securities involves a high degree of risk. See “Risk Factors” beginning on page 11 of this prospectus for
a discussion of information that should be considered in connection with an investment in our securities.
Neither
the Securities and Exchange Commission (“SEC”) nor any state securities commission has approved or disapproved of these securities
or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
We
are an “emerging growth company” as that term is used in the Jumpstart Our Business Startups Act of 2012, and we have elected
to comply with certain reduced public company reporting requirements.
| | |
Per
Share
And
Accompanying
Common
Warrant | | |
Per
Pre-funded
Warrant and
Accompanying
Common
Warrant | | |
Total | |
| Public offering price | |
$ | | | |
$ | | | |
$ | | |
| Placement agent fees(1) | |
$ | | | |
$ | | | |
$ | | |
| Proceeds, before expenses,
to us(2) | |
$ | | | |
$ | | | |
$ | | |
| (1) |
Represents a cash fee equal
to [*]% of the aggregate purchase price paid by investors in this offering. See “Plan of Distribution” beginning
on page 112 of this prospectus for a description of the compensation to be received by the placement agent. |
| |
|
| (2) |
The amount of offering
proceeds to us presented in this table does not give effect to any exercise of the Common Warrants or Pre-Funded Warrants. |
We
anticipate that delivery of the securities against payment therefor will be made on or before ,
2024.
The
date of this prospectus is , 2024.
TABLE
OF CONTENTS
You
should rely only on the information contained in this prospectus or any prospectus supplement or amendment. Neither we, nor the placement
agent, have authorized any other person to provide you with information that is different from, or adds to, that contained in this prospectus.
If anyone provides you with different or inconsistent information, you should not rely on it. Neither we nor the placement agent take
responsibility for, and can provide no assurance as to the reliability of, any other information that others may give you. You should
assume that the information contained in this prospectus, or any free writing prospectus is accurate only as of the date of this prospectus,
regardless of the time of delivery of this prospectus or of any sale of our common stock. Our business, financial condition, results
of operations and prospects may have changed since that date. We are not making an offer of any securities in any jurisdiction in which
such offer is unlawful.
No
action is being taken in any jurisdiction outside the United States to permit a public offering of our securities or possession or distribution
of this prospectus in that jurisdiction. Persons who come into possession of this prospectus in jurisdictions outside the United States
are required to inform themselves about and to observe any restrictions as to this public offering and the distribution of this prospectus
applicable to that jurisdiction.
ABOUT
THIS PROSPECTUS
Throughout
this prospectus, unless otherwise designated or the context suggests otherwise,
| ● | all
references to the “Tenon,” the “Company,” the “registrant,”
“we,” “our,” or “us” in this prospectus mean Tenon Medical,
Inc.; |
| ● | “year”
or “fiscal year” means the year ending December 31st; and |
| ● | all
dollar or $ references, when used in this prospectus, refer to United States dollars. |
Market
Data
Market
data and certain industry data and forecasts used throughout this prospectus were obtained from internal company surveys, market research,
consultant surveys, publicly available information, reports of governmental agencies and industry publications and surveys. Industry
surveys, publications, consultant surveys and forecasts generally state that the information contained therein has been obtained from
sources believed to be reliable, but the accuracy and completeness of such information is not guaranteed. To our knowledge, certain third-party
industry data that includes projections for future periods does not consider the effects of the worldwide coronavirus pandemic. Accordingly,
those third-party projections may be overstated and should not be given undue weight. We have not independently verified any of the data
from third party sources, nor have we ascertained the underlying economic assumptions relied upon therein. Similarly, internal surveys,
industry forecasts and market research, which we believe to be reliable based on our management’s knowledge of the industry, have
not been independently verified. Forecasts are particularly likely to be inaccurate, especially over long periods of time. In addition,
we do not necessarily know what assumptions regarding general economic growth were used in preparing the forecasts we cite. Statements
as to our market position are based on the most currently available data. While we are not aware of any misstatements regarding the industry
data presented in this prospectus, our estimates involve risks and uncertainties and are subject to change based on various factors,
including those discussed under the heading “Risk Factors” in this prospectus. We are, however, liable for the information
in the prospectus related to the market and industry data.
PROSPECTUS
SUMMARY
This
summary provides a brief overview of the key aspects of our business and our securities. The reader should read the entire prospectus
carefully, especially the risks of investing in our common stock discussed under “Risk Factors.” Some of the statements contained
in this prospectus, including statements under “Summary” and “Risk Factors” as well as those noted in the documents
incorporated herein by reference, are forward-looking statements and may involve a number of risks and uncertainties. Our actual results
and future events may differ significantly based upon a number of factors. The reader should not put undue reliance on the forward-looking
statements in this document, which speak only as of the date on the cover of this prospectus.
Introduction
The
Company was incorporated in the State of Delaware on June 19, 2012 and was headquartered in San Ramon, California until June 2021 when
it relocated to Los Gatos, California. The Company is a medical device company that has developed The Catamaran™ SI Joint Fusion
System (“The Catamaran System”) that offers a novel, less invasive approach to the sacroiliac joint (the “SI Joint”)
using a single, robust, titanium implant for treatment of the most common types of SI Joint disorders that cause lower back pain. The
Company received U.S. Food and Drug Administration (“FDA”) clearance in 2018 for The Catamaran System and is currently focused
on the US market. Since the national launch of The Catamaran System in October 2022, the Company is focused on three commercial opportunities:
1) Primary SI Joint procedures, 2) Revision procedures of failed SI Joint implants and 3) SI Joint fusion adjunct to a spine fusion construct.
The
Opportunity
We
estimate that over 30 million American adults have chronic lower back pain. Published clinical studies have shown that 15% to 30% of
all chronic lower back pain is associated with the SI-Joint. For patients whose chronic lower back pain stems from the Sacroiliac Joint
(“SI-Joint”), our experience in both clinical trials and commercial settings indicates the system to be introduced by Tenon
could be beneficial for patients who are properly diagnosed and screened for surgery by trained healthcare providers.
In
2019, approximately 475,000 patients in the United States were estimated to have received an aesthetic injection to temporarily alleviate
pain emanating from the SI-Joint and/or to diagnose SI-Joint pain. Additionally, several non-surgical technologies have been introduced
in the past 10 years to address patients who do not respond to conservative options, including systemic oral medications, opioids, physical
therapy and injection therapy.
To
date, the penetration of a surgical solution for this market has been relatively low (5-7%). We believe this is due to complex surgical
approaches and suboptimal implant design of existing options. The penetration of this market with an optimized surgical solution is Tenon’s
focus.
We
believe the SI-Joint is the last major joint to be successfully addressed by the spine implant industry. Studies have shown that disability
resulting from disease of the SI-Joint is comparable to the disability associated with a number of other serious spine conditions, such
as knee and hip arthritis and degenerative disc disease, each of which has surgical solutions where an implant is used, and a multi-billion-dollar
market exists.
The SI-Joint
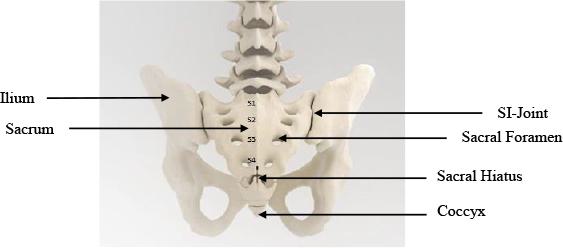
The
SI-Joint is a strong weight bearing synovial joint situated between the lumbar spine and the pelvis and is aligned along the longitudinal
load bearing axis of the human spine when in an upright posture. It functions as a force transfer conduit where it transfers axial loads
bi-directionally from the spine to the pelvis and lower extremities and allows forces to be transmitted from the extremities to the spine.
It also provides load sharing between the hip and spine to contribute towards attenuation of impact shock and stress from activities
of daily living.
The
SI-Joint is a relatively immobile joint that connects the sacrum (the spinal segment that is attached to the base of the lumbar spine
at the L5 vertebra) and the ilium of the pelvis. Each SI-Joint is approximately 2-4mm wide and irregularly shaped.
Motion
of the SI-Joint features vertical shear and rotation. Although the rotational forces about the SI-Joint are relatively low, repetitive
motions created by daily activities such as walking, jogging, twisting at the hips, and jumping can increase the stresses on the SI-Joint.
If the SI-Joint is compromised through injury or degeneration, the load bearing and motion restraints from the surrounding anatomical
structures of the SI-Joint will be compromised resulting in abnormal stress transfers across the joint to these structures, thereby further
augmenting the degenerative cascade of the SI-Joint. Eventual pain and cessation of an individual’s normal activities due to a
painful and unstable SI-Joint have led to an increase in the recent development of SI-Joint stabilization devices.
Non-Surgical
Treatment of Sacroiliac Joint Disease
Several
non-surgical treatments exist for suspected sacroiliac joint pain. These conservative steps often provide desired relief for the patient.
Non-surgical treatments include:
| ● | Drug
Therapy: including opiates and non-steroidal anti-inflammatory medications. |
| ● | Intra-Articular
Injections of Steroid Medications: which are typically performed by physicians who specialize
in pain treatment or anesthesia. |
| ● | Radiofrequency
Ablation: or the cauterizing of the lateral branches of the sacral nerve roots. |
When
conservative steps fail to deliver sustained pain relief and return to quality of life, specific diagnostic protocols are utilized to
explore if a surgical option should be considered.
Diagnosis
Historically,
diagnosing pain from the SI-Joint was not routinely a focus of orthopedic or neurosurgery training during medical school or residency
programs. Due to its invasiveness, post-operative pain, and muscle disruption along with a difficult procedure overall, the open SI-Joint
fusion procedure was rarely taught in these settings.
The
emergence of various SI-Joint surgical technologies has generated a renewed discussion of SI-Joint issues. Of particular focus is the
diagnostic protocol utilized to properly select patients for SI-Joint surgery. Patients with low back pain typically start with primary
care physicians who often refer to pain specialists. Here, the patient will undergo traditional physical therapy combined with oral medications
(anti-inflammatory, narcotic, etc.). If the patient fails to respond to these steps the pain specialist may move to therapeutic injections
of the SI-Joint. These injections may serve to lessen inflammation to the point that the patient is satisfied. However, the impact from
these injections is often transient. In this case the patient is often referred to a clinician to determine if the patient may be a candidate
for surgical intervention. A series of provocative tests in clinic, combined with a specific injection protocol to isolate the SI-Joint
as the pain generator is then utilized to confirm the need for surgical intervention. Published literature has shown this technique to
be a very effective step to determine the best treatment to alleviate pain.
Limitations
of Existing Treatment Options
Surgical
fixation and fusion of the SI-Joint with an open surgical technique was first reported in 1908, with further reports in the 1920s. The
open procedure uses plates and screws, requires a 6 to 12-inch incision and is extremely invasive. Due to the high invasiveness and associated
morbidity, the use of this procedure is limited to cases involving significant trauma, tumor, etc.
Less
invasive surgical options along with implant design began to emerge over the past 15 years. These options feature a variety of approaches
and implant designs and have been met with varying degrees of adoption. Lack of a standard and accepted diagnostic approach, complexity
of approach, high morbidity of approach, abnormally high complication rates and inability to radiographically confirm fusion have all
been cited as reasons for low adoption of these technologies.
Commercialization
Tenon
initiated its national commercial launch of The Catamaran System in October 2022 to address what we believe is a large market opportunity.
The Catamaran System includes instruments and implants designed to prepare and fixate the SI-Joint for fusion. The Catamaran System is
distinct from other competitive offerings in the following ways:
| ● | Inferior
/ Posterior Sacroiliac Fusion Approach |
| ● | Reduced
Approach Morbidity |
| ● | Direct
And Visualized Approach to the SI-Joint |
| ● | Single
Implant Technique |
| ● | Insertion
Trajectory Away from the Neural Foramen |
| ● | Insertion
Trajectory Away from Major Lateral Vascular Structures |
| ● | Autologous
Bone Grafting in the Ilium, Sacrum and Bridge |
| ● | Radiographic
Confirmation of Bridging Bone Fusion of the SI-Joint |
The
fixation device and its key features are shown below:
 |
Key
Features
“Pontoon”
in the ilium
“Pontoon”
in the sacrum
“Pontoons
and Bridge” filled with autologous bone from drilling process
Leading
edge osteotome creates defect and facilitates ease of insertion |
The
Catamaran System is a singular implant designed with several proprietary components which allow for it to be explicitly formatted to
address the SI-Joint with a single approach and implant. This contrasts with several competitive implant systems that require multiple
approach pathways and implants to achieve fixation. In addition, the inferior-posterior approach is designed to be direct to the joint
and through limited anatomical structures which may minimize the morbidity of the approach. The implant features a patented dual pontoon
open cell design which enables the clinician to pack the pontoons with the patient’s own autologous bone designed to promote bone
fusion across the joint. The Catamaran System is designed specially to resist vertical shear and rotation of the joint in which it was
implanted, helping stabilize the joint in preparation for eventual fusion.
The
instruments we have developed are proprietary to The Catamaran System and specifically designed to transfix the SI-Joint and facilitate
an inferior-posterior approach that is unique to the system.
Tenon
also has developed a proprietary 2D placement protocol as well as a protocol for 3D navigation utilizing the latest techniques in spine
surgery. These Tenon advancements are intended to further enhance the safety of the procedure and encourage more physicians to adopt
the procedure.
In
October 2022, we received Institutional Review Board (“IRB”) approval from WCG IRB for two separate Tenon-sponsored post
market clinical studies of The Catamaran System. The approval by WCG allows designated Catamaran study centers to begin recruiting and
enrolling patients into the clinical studies. The first approval from WCG IRB will support a prospective, multi-center, single arm post
market study that will evaluate the clinical outcomes of patients with sacroiliac joint disruptions or degenerative sacroiliitis treated
with The Catamaran System. Patients will be followed out to 24 months assessing various patient reported outcomes, radiographic assessments,
and adverse events. The second prospective, multi-center, Catamaran study will evaluate 6-to-12-month radiographic outcomes to assess
fusion of patients that have already undergone treatment with The Catamaran System. In addition, retrospective and prospective clinical
outcomes will be evaluated. We anticipate completing enrollment by the end of the third quarter of 2024.
For
a description of the challenges, we face and the risks and limitations that could harm our prospects, see “Risk Factors.”
Recent
Developments
Increase
of shares subject to our Incentive Stock Option Plan
On
July 23, 2024, at our annual meeting of stockholders, our stockholders approved a 1.1 million share increase in the number of shares
subject to the Tenon Medical, Inc. 2022 Equity Incentive Plan (the “2022 Plan”). There are currently 1,408,959 shares reserved
for issuance under the 2022 Plan, of which 1,154,173 shares are available for future grants.
Retirement
of the Company’s Chief Financial Officer
On
July 31, 2024, Steven Van Dick retired from his positions as Executive Vice President, Finance and Administration, Chief Financial Officer
and Assistant Secretary. The Company is actively searching for a qualified candidate to fill the role of Chief Financial Officer. Mr.
Van Dick will continue as a part time advisor to the Company and continue to assist with the transition of his responsibilities and provide
strategic advice as requested until December 31, 2024.
Nasdaq
Notice of Failure to Comply with Continued Listing Standards
On
May 7, 2024, we received a letter from the Nasdaq Listing Qualifications Staff of The Nasdaq Stock Market LLC (“Nasdaq”)
stating that for the 30 consecutive business day period between March 25, 2024 and May 6, 2024, our common stock had not maintained a
minimum closing bid price of $1.00 per share required for continued listing on The Nasdaq Capital Market pursuant to Nasdaq Listing Rule
5550(a)(2) (the “Bid Price Rule”). Pursuant to Nasdaq Listing Rule 5810(c)(3)(A), we were provided an initial period of 180
calendar days, or until November 4, 2024 (the “Compliance Period”), to regain compliance with the Bid Price Rule.
To
regain compliance, the closing bid price of our common stock must meet or exceed $1.00 per share for a minimum of 10 consecutive trading
days, unless extended by Nasdaq under Nasdaq Rule 5810(c)(3)(H), prior to November 4, 2024.
If
we do not regain compliance with the Bid Price Rule by November 4, 2024, we may be eligible for an additional 180-day period to regain
compliance if we meet all of the other Nasdaq listing criteria and if Nasdaq does not believe we will not be able to regain compliance
within such 180-day period. If we cannot regain compliance during the Compliance Period or any subsequently granted compliance period,
our common stock will be subject to delisting.
Our
common stock continues to be listed on The Nasdaq Capital Market under the symbol “TNON”. We are currently evaluating our
options for regaining compliance.
The
notice from Nasdaq has no immediate effect on the listing or trading of our common stock on The Nasdaq Capital Market and does not affect
our business, operations or reporting requirements with the SEC.
On
December 21, 2023, at a special meeting of our stockholders our stockholders approved an amendment to our Certificate of Incorporation
, at our annual meeting of stockholders, our stockholders approved to provide for a reverse stock split (the “Reverse Stock Split”)
of the Common Stock, that will be at a ratio ranging from one for two (1:2) to one for fifty (1:50), the final determination of which
shall be determined by the Board. We may effect the Reverse Stock split within the approved range to regain compliance with the Bid Price
Rule.
Exchange
Offer
On
April 8, 2024, we launched a one-time stock option exchange program (the “Option Exchange”) pursuant to which eligible participants
were able to exchange outstanding stock options for a lesser amount of new restricted stock units (“RSUs”). Our executive
officers, non-employee directors and consultants were eligible to participate in the Option Exchange. Employees, non-employee directors
and consultants received one RSU for every two shares of our common stock underlying the eligible options surrendered. This “exchange
ratio” (2-for-1) was applied on a grant-by-grant basis. The Option Exchange expired on May 6, 2024 at 11:59 p.m., Eastern Time.
At that time, stock options to purchase 83,391 shares of our common stock were surrendered and 41,698 new RSUs were issued under the
2022 Plan.
2024
Series A Offering
On
February 20, 2024, we entered into a Securities Purchase Agreement (the “Series A Purchase Agreement”) with certain investors
(the “Series A Investors”), pursuant to which the Company agreed to sell, issue and deliver to the Series A Investors, in
a private placement offering (the “Series A Offering”), a total of 172,239 shares of the Company’s Series A Preferred
Stock (the “Series A Preferred Stock”) and warrants (the “Series A Warrants”) to purchase 258,374 shares of common
stock, par value $0.001 per share, of the Company (“Common Stock”) at an exercise price equal to $1.2705 per share for an
aggregate offering price of $2,605,000. Under the Series A Purchase Agreement, each Series A Investor paid $15.125 for each share of
Series A Preferred Stock. In addition, each investor received Series A Warrants to purchase a number of shares of our common stock equal
to 15% of the number of shares of our common stock underlying the shares of Series A Preferred Stock purchased by such investor. In connection
with the offering of the Series A Preferred Stock the Company exchanged the Notes (as defined below) for 84,729 shares of Series A Preferred
Stock and Series A Warrants to purchase 157,094 shares of our common stock. There are a total of 256,968 shares of Series A Preferred
Stock outstanding as of August 13, 2024.
2023
Note Offering
On
November 21, 2023, we entered into securities purchase agreements with certain investors (the “Note Investors”), pursuant
to which we agreed to sell, issue and deliver to the Note Investors, in a private placement offering (the “Note Offering”),
a total of $1,250,000 in secured notes (the “Notes”) and warrants (the “Note Warrants”) to purchase 45,000 shares
of our common stock at an exercise price equal to $1.94 per share. The Company received $1,125,000 from the Note Offering after payment
of investor expenses. As described above, the Notes have been repaid in full and are no longer outstanding.
The
Note Warrants expire five (5) years from the issuance date of the Note Warrants. The Note Warrants contain a “cashless exercise”
feature and contain anti-dilution rights on subsequent issuances of equity or equity equivalents.
Summary
Risk Factors
Our
business is subject to numerous risks and uncertainties, any one of which could materially adversely affect our results of operations,
financial condition or business. These risks include, but are not limited to, those listed below. This list is not complete, and should
be read together with the section titled “Risk Factors” below:
| ● | We
have incurred losses in the past, our financial statements have been prepared on a going
concern basis and we may be unable to achieve or sustain profitability in the future; |
| ● | Practice
trends or other factors, may cause procedures to shift from the hospital environment to ambulatory
surgical centers (“ASCs”), where pressure on the prices of our products is generally
more acute; |
| ● | If
hospitals, clinicians, and other healthcare providers are unable to obtain and maintain coverage
and reimbursement from third-party payors for procedures performed using our products, adoption
of our products may be delayed, and it is unlikely that they will gain further acceptance; |
| ● | We
may not be able to convince physicians that The Catamaran System is an attractive alternative
to our competitors’ products and that our procedure is an attractive alternative to
existing surgical and non-surgical treatments of the SI-Joint; |
| ● | Clinicians
and payors may not find our clinical evidence to be compelling, which could limit our sales,
and ongoing and future research may prove our products to be less safe and effective than
initially anticipated; |
| ● | Pricing
pressure from our competitors, changes in third-party coverage and reimbursement, healthcare
provider consolidation, payor consolidation and the proliferation of “physician-owned
distributorships” may impact our ability to sell our product at prices necessary to
support our current business strategies; |
| ● | We
operate in a very competitive business environment and if we are unable to compete successfully
against our existing or potential competitors, our sales and operating results may be negatively
affected and we may not grow; |
| ● | We
currently manufacture (through third parties) and sell products used in a single procedure,
which could negatively affect our operations and financial condition; |
| ● | Our
sales volumes and our operating results may fluctuate over the course of the year; |
| ● | Various
factors outside our direct control may adversely affect manufacturing and distribution of
our product; |
| ● | We
are dependent on a limited number of contract manufacturers, some of them single-source and
some of them in single locations, for our product, and the loss of any of these contract
manufacturers, or their inability to provide us with an adequate supply of products in a
timely and cost-effective manner, could materially adversely affect our business; |
| ● | As
our sales grow, our contract manufacturers may encounter problems or delays in the manufacturing
of our product or fail to meet certain regulatory requirements which could result in an adverse
effect on our business and financial results; |
| ● | The
size and future growth in the market for the SI-Joint fixation market have not been established
based on market reports and our estimates are based on our own review and analysis of public
information and may be smaller than we estimate, possibly materially. In addition, our estimates
of cost savings to the economy and healthcare system as a result of The Catamaran System
procedure are based on our internal estimates and market research and could also be smaller
than we estimate, possibly materially. If our estimates and projections overestimate the
size of this market or cost savings, our sales growth may be adversely affected; |
| ● | If
we experience significant disruptions in our information technology systems, our business,
results of operations, and financial condition could be adversely affected; |
| ● | We
may seek to grow our business through acquisitions of or investments in new or complementary
businesses, products or technologies, and the failure to manage acquisitions or investments,
or the failure to integrate them with our existing business, could have a material adverse
effect on us; |
| ● | We
may enter into collaborations, in-licensing arrangements, joint ventures, strategic alliances,
or partnerships with third-parties that may not result in the development of commercially
viable products or the generation of significant future revenue; |
| ● | We
are increasingly dependent on information technology, and our systems and infrastructure
face certain risks, including cybersecurity and data leakage risks; |
| ● | Geopolitical
conditions, including trade disputes and direct or indirect acts of war or terrorism, could
have an adverse effect on our operations and financial results; |
| ● | Inflation
may adversely affect our operations and financial results; |
| ● | We
and our contract manufacturers are subject to extensive governmental regulation both in the
United States and abroad, and failure to comply with applicable requirements could cause
our business to suffer; |
| ● | Our
employees, independent contractors, consultants, contract manufacturers, and our independent
sales representatives may engage in misconduct or other improper activities, relating to
regulatory standards and requirements; |
| ● | We
are subject to environmental laws and regulations that can impose significant costs and expose
us to potential financial liabilities; |
| ● | Our
ability to protect our intellectual property and proprietary technology is uncertain; |
| ● | We
may not be able to protect our intellectual property rights throughout the world; |
| ● | The
sale or issuance of our common stock to Lincoln Park may cause dilution and the sale of the
shares of common stock acquired by Lincoln Park, or the perception that such sales may occur,
could cause the price of our common stock to fall; and |
| ● | Our
management will have broad discretion over the use of the net proceeds from our sale of shares
of common stock to Lincoln Park, and you may not agree with how we use the proceeds and the
proceeds may not be invested successfully. |
Corporate
Information
Our
principal executive offices are located at 104 Cooper Court, Los Gatos, CA 95032. Our website address is www.tenonmed.com. The information
included on our website is not part of this prospectus.
Implications
of Being an Emerging Growth Company
We
are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012 (the “JOBS Act”).
We will remain an emerging growth company until the earlier of (i) the last day of the fiscal year following the fifth anniversary of
the date of the first sale of our common stock pursuant to an effective registration statement under the Securities Act; (ii) the last
day of the fiscal year in which we have total annual gross revenues of $1.235 billion or more; (iii) the date on which we have issued
more than $1 billion in nonconvertible debt during the previous three years; or (iv) the date on which we are deemed to be a large accelerated
filer under applicable SEC rules. We expect that we will remain an emerging growth company for the foreseeable future, but cannot retain
our emerging growth company status indefinitely and will no longer qualify as an emerging growth company on or before the last day of
the fiscal year following the fifth anniversary of the date of the first sale of our common stock pursuant to an effective registration
statement under the Securities Act. For so long as we remain an emerging growth company, we are permitted and intend to rely on exemptions
from specified disclosure requirements that are applicable to other public companies that are not emerging growth companies.
These
exemptions include:
| ● | being
permitted to provide only two years of audited financial statements, in addition to any required
unaudited interim financial statements, with correspondingly reduced “Management’s
Discussion and Analysis of Financial Condition and Results of Operations” disclosure; |
| ● | not
being required to comply with the requirement of auditor attestation of our internal controls
over financial reporting; |
| ● | not
being required to comply with any requirement that may be adopted by the Public Company Accounting
Oversight Board regarding mandatory audit firm rotation or a supplement to the auditor’s
report providing additional information about the audit and the financial statements; |
| ● | reduced
disclosure obligations regarding executive compensation; and |
| ● | not
being required to hold a nonbinding advisory vote on executive compensation and shareholder
approval of any golden parachute payments not previously approved. |
We
have taken advantage of certain reduced reporting requirements in this prospectus. Accordingly, the information contained herein may
be different than the information you receive from other public companies in which you hold stock.
An
emerging growth company can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act for
complying with new or revised accounting standards. This allows an emerging growth company to delay the adoption of certain accounting
standards until those standards would otherwise apply to private companies. We have irrevocably elected to avail ourselves of this extended
transition period and, as a result, we will not be required to adopt new or revised accounting standards on the dates on which adoption
of such standards is required for other public reporting companies.
We
are also a “smaller reporting company” as defined in Rule 12b-2 of the Securities Exchange Act of 1934, as amended (the “Exchange
Act”), and have elected to take advantage of certain of the scaled disclosure available for smaller reporting companies.
SUMMARY
OF THE OFFERING
| Securities
offered by us |
|
Up
to [*] shares of common stock and accompanying Common Warrants to purchase up to [*] shares of common stock at a assumed combined
public offering price of $[*] per share and Common Warrant, based on the closing price of our common stock on Nasdaq, on [*], 2024.
Each Warrant will have an exercise price of $[*] per share (100% of the combined public offering price per share and accompanying
Common Warrant), is exercisable immediately and will expire five years from the date of issuance. Each share of common stock and
accompanying Common Warrant is immediately separable upon issuance and will be issued separately in this offering. |
| |
|
|
| Pre-Funded
Warrants offered by us |
|
We are also offering to
those purchasers whose purchase of common stock in this offering would otherwise result in the purchaser, together with its affiliates
and certain related parties, beneficially owning more than 4.99% (or, at the election of the purchaser, 9.99%) of our outstanding
common stock immediately following the closing of this offering, in lieu of purchasing common stock, Pre-funded Warrants to purchase
up to an aggregate of [*] shares of our common stock. Each Pre-funded Warrant is exercisable for one share of our common stock. The
purchase price of each Pre-funded Warrant is equal to the price at which a share of common stock is being sold to the public in this
offering, minus $0.0001, and the exercise price of each Pre-funded Warrant is $0.0001 per share. The Pre-funded Warrants are exercisable
immediately and may be exercised at any time until all of the Pre-funded Warrants are exercised in full. This offering also relates
to the shares of common stock issuable upon exercise of any Pre-funded Warrants sold in this offering. For each Pre-funded Warrant
that we sell, the number of shares of common stock that we are offering will be reduced on a one-for-one basis |
| |
|
|
| Common
Warrants offered by us |
|
Each share of common stock
will be sold together with one Common Warrant. Each Common Warrant has an exercise price per share equal to 100% of the combined
public offering price per share and accompanying Common Warrant. the Common Warrant is exercisable immediately and will expire
five years from the date of issuance. Because we will issue a Common Warrant for each share of common stock and for each Pre-funded
Warrant sold in this offering, the number of Common Warrants sold in this offering will not change as a result of a change in the
mix of shares of Common Stock and Pre-Funded Warrants sold. See “Description of the Securities–Warrants”. |
| |
|
|
| Common
stock to be outstanding after the offering(1)(2) |
|
[*] shares (assuming no
exercise of the Common Warrants and no sale of Pre-funded Warrants). |
| Use of Proceeds |
|
We currently
intend to use the net proceeds to us from this offering to expand the commercial launch of our product including training clinicians
on The CATAMARAN System procedure, continuing clinical marketing studies that are focused on capturing post-market safety data, hire
additional employees, other marketing activities and for working capital and general corporate purposes. See the section of this
prospectus titled “Use of Proceeds” beginning on page 46. |
| |
|
|
| Listing |
|
Our
common stock and tradeable warrants (“Tradeable Warrants”) trade on The Nasdaq Capital Market under the symbols “TNON”
and “TNONW,” respectively.
There
is no established public trading market for the Warrants and Pre-Funded Warrants and we do not expect a market to develop. In addition,
we do not intend to apply to list the Pre-Funded Warrants on any national securities exchange or other nationally recognized trading
system. Without an active trading market, the liquidity of the Pre-Funded Warrants will be limited. |
| |
|
|
| Risk Factors |
|
You should carefully consider
the information set forth in this prospectus and, in particular, the specific factors set forth in the “Risk Factors”
section beginning on page 11 of this prospectus before deciding whether or not to invest in shares of our common stock. |
| |
|
|
| Transfer Agent and registrar |
|
VStock Transfer, LLC. |
| |
|
|
| Reasonable best efforts |
|
We have agreed to offer
and sell the securities offered hereby to the purchasers through the placement agent. The placement agent is not required to buy
or sell any specific number or dollar amount of the securities offered hereby, but it will use its reasonable best efforts to solicit
offers to purchase the securities offered by this prospectus. See “Plan of Distribution” on page 112 of this prospectus. |
| |
|
|
| Lock-up agreements |
|
We
have agreed for a period of [*] ([*]) days after the closing date not to [*].
Each
of our executive officers and directors have agreed with the placement agent not to [*]. For additional information regarding our
arrangement with the placement agent, please see “Plan of Distribution.” |
| (1) | The
number of shares of common stock to be outstanding after this offering is based on 3,951,767
shares of common stock outstanding as of August 13, 2024, and excludes: |
| ● | 256,968 shares of our common stock underlying our Series A Preferred
Stock, which are convertible into 2,569,680 shares of common stock and vote with the common stock at a ratio of 10 votes for every one
share of Series A Preferred Stock; |
| ● | 45,000
shares of our common stock underlying our Note Warrants, with a per share exercise price
equal to the offering price of one share of common stock and the accompanying Common Warrant; |
| ● | 415,468 shares of our common stock underlying our Series A Warrants,
with a per share exercise price equal to $1.2705; |
| ● | 72,563 shares of our common stock issuable pursuant to granted equity
awards per our equity incentive plan, with a weighted average exercise price of $3.12 per share; |
| ● | 136,690
shares of our common stock issuable pursuant to restricted stock units granted pursuant to
our equity incentive plan; and |
| ● | 9,600
shares of our common stock issuable upon the exercise of warrants issued to the underwriters
in our initial public offering that closed on April 29, 2022, with an exercise price of $50.00
per share. |
| (2) | Unless
otherwise indicated, this prospectus reflects and assumes the following: |
| ● | no
exercise of outstanding options or warrants described above; and |
| ● | no
exercise of the Common Warrants, and no sale of Pre-funded Warrants, which, if sold, would
reduce the number of shares of common stock that we are offering on a one-for-one basis. |
RISK
FACTORS
Our
business is subject to many risks and uncertainties, which may affect our future financial performance. If any of the events or circumstances
described below occur, our business and financial performance could be adversely affected, our actual results could differ materially
from our expectations, and the price of our stock could decline. The risks and uncertainties discussed below are not the only ones we
face. There may be additional risks and uncertainties not currently known to us or that we currently do not believe are material that
may adversely affect our business and financial performance. You should carefully consider the risks described below, together with all
other information included in this prospectus including our financial statements and related notes, before making an investment decision.
The statements contained in this prospectus that are not historic facts are forward-looking statements that are subject to risks and
uncertainties that could cause actual results to differ materially from those set forth in or implied by forward-looking statements.
If any of the following risks actually occurs, our business, financial condition or results of operations could be harmed. In that case,
the trading price of our common stock could decline, and investors in our securities may lose all or part of their investment.
Risks
Related to Our Business and Operations
We
have incurred losses in the past, our financial statements have been prepared on a going concern basis and we may be unable to achieve
or sustain profitability in the future.
To
date, we have financed our operations primarily through the issuance of public and private equity and convertible notes. We have devoted
substantially all of our resources to research and development, creating the infrastructure for a publicly traded medical device company,
preparing for our national commercial launch, and clinical and regulatory matters for our products. There can be no assurances that we
will be able to generate sufficient revenue from our existing products or from any future product candidates to transition to profitability
and generate consistent positive cash flows. We expect that our operating expenses will continue to increase as we continue to build
our commercial infrastructure, develop, enhance, and commercialize our existing and new products and incur additional operating and reporting
costs associated with being a public company. As a result, we expect to continue to incur operating losses for the foreseeable future
and may never achieve profitability. Furthermore, even if we do achieve profitability, we may not be able to sustain or increase profitability
on an ongoing basis. If we do not achieve profitability, it will be more difficult for us to finance our business and accomplish our
strategic objectives.
Our
recurring losses from operations and negative cash flows raise substantial doubt about our ability to continue as a going concern. As
a result, our independent registered public accounting firm included an explanatory paragraph in its report on our financial statements
for the fiscal year ended, December 31, 2023, describing the existence of substantial doubt about our ability to continue as a going
concern. Our expected future capital requirements may depend on many factors including expanding our clinician base, increasing the rate
at which we train clinicians, the number of additional clinical papers initiated, and the timing and extent of spending on the development
of our technology to increase our product offerings. We may need additional funding to fund our operations but additional funds may not
be available to us on acceptable terms on a timely basis, if at all. We may seek funds through borrowings or through additional rounds
of financing, including private or public equity or debt offerings. If we raise additional funds by issuing equity securities, our stockholders
may experience dilution. Any future debt financing into which we enter may impose upon us additional covenants that restrict our operations,
including limitations on our ability to incur liens or additional debt, pay dividends, repurchase our common stock, make certain investments,
and engage in certain merger, consolidation or asset sale transactions. Any future debt financing or additional equity that we raise
may contain terms that are not favorable to us or our stockholders. Furthermore, we cannot be certain that additional funding will be
available on acceptable terms, if at all. If we are unable to raise additional capital or generate sufficient cash from operations to
adequately fund our operations, we will need to curtail planned activities to reduce costs, which will likely harm our ability to execute
on our business plan and continue operations.
If
hospitals, clinicians, and other healthcare providers are unable to obtain coverage and reimbursement from third-party payors for procedures
performed using our products, adoption of our products may be delayed, and it is unlikely that they will gain further acceptance.
Growing
sales of our product depends on the availability of adequate coverage and reimbursement from third-party payors, including government
programs such as Medicare and Medicaid, private insurance plans, and managed care programs. Hospitals, clinicians, and other healthcare
providers that purchase or use medical devices generally rely on third-party payors to pay for all or part of the costs and fees associated
with the procedures performed with these devices.
Adequate
coverage and reimbursement for procedures performed with our products is central to the acceptance of our current and future products.
We may be unable to sell our products on a profitable basis if third-party payors deny coverage, continue to deny coverage or reduce
their current levels of payment, or if our costs for the product increase faster than increases in reimbursement levels.
Many
private payors refer to coverage decisions and payment amounts determined by the Centers for Medicare and Medicaid Services, or CMS,
which administers the Medicare program, as guidelines for setting their coverage and reimbursement policies. By June 30, 2016, all Medicare
Administrative Contractors were regularly reimbursing for minimally invasive and/or open SI-Joint fusion. Private payors that do not
follow the Medicare guidelines may adopt different coverage and reimbursement policies for procedures performed with our products. Private
commercial payors have been slower to adopt positive coverage policies for minimally invasive and/or open SI-Joint fusion, and many private
payors still have policies that treat the procedure as experimental or investigational and do not regularly reimburse for the procedure.
Future action by CMS or third-party payors may further reduce the availability of payments to physicians, outpatient surgery centers,
and/or hospitals for procedures using our products.
The
healthcare industry in the United States has experienced a trend toward cost containment as government and private insurers seek to control
healthcare costs. Payors are imposing lower payment rates and negotiating reduced contract rates with service providers and being increasingly
selective about the technologies and procedures they choose to cover. There can be no guarantee that we will be able to provide the scientific
and clinical data necessary to overcome these policies. Payors may adopt policies in the future restricting access to medical technologies
like ours and/or the procedures performed using such technologies. Therefore, we cannot be certain that the procedures performed with
each of our products will be reimbursed. There can be no guarantee that, should we introduce additional products in the future, payors
will cover those products or the procedures in which they are used.
If
the reimbursement provided by third-party payors to hospitals, clinicians, and other healthcare providers for procedures performed using
our products is insufficient, adoption and use of our products and the prices paid for our implants may decline.
When
a Tenon procedure utilizing The Catamaran System is performed, both the clinician and the healthcare facility, a hospital (inpatient
or outpatient clinic), submit claims for reimbursement to the patient’s insurer. Generally, the facility obtains a lump sum payment,
or facility fee, for SI-Joint fusions. Our products are purchased by the facility, along with other supplies used in the procedure. The
facility must also pay for its own fixed costs of operation, including certain operating room personnel involved in the procedure, and
other medical services care. If these costs exceed the facility reimbursement, the facility’s managers may discourage or restrict
clinicians from performing the procedure in the facility or using certain technologies, such as The Catamaran System, to perform the
procedure.
The
Medicare 2022 national average hospital inpatient payment ranges from approximately $25,000 to approximately $59,000 depending on the
procedural approach and the presence of Complication and Comorbidity (CC)/Major Complication and Comorbidity (MCC).
The
Medicare 2022 national average hospital outpatient clinic payment is $21,897. We believe that insurer payments to facilities are generally
adequate for these facilities to offer The Catamaran System. However, there can be no guarantee that these facility payments will not
decline in the future. The number of procedures performed, and the prices paid for our implants may in the future decline if payments
to facilities for SI-Joint fusions decline.
Clinicians
are reimbursed separately for their professional time and effort to perform a surgical procedure. Depending on the surgical approach,
the incision size, type and extent of imaging guidance, indication for procedure, and the insurer, The Catamaran System procedure may
be reported by the clinician using any one of the applicable following CPT® codes 27279, 27280, 27299. The Medicare 2022 national
average payment for CPT® 27279 is $807 and $1,325 for 27280. CPT® 27299 has no national valuation. Clinicians, however, can present
a crosswalk to another procedure believed to be fairly equivalent and/or comparison to a code for which there is an existing valuation.
For
some governmental programs, such as Medicaid, coverage and reimbursement differ from state to state, and some state Medicaid programs
may not pay an adequate amount for the procedures performed with our products, if any payment is made at all. Similar to Medicaid, many
private payors’ coverage and payment may differ from one payer to another as well.
We
believe that some clinicians view the current Medicare reimbursement amount as insufficient for the procedure, given the work effort
involved with the procedure, including the time to diagnose the patient and obtain prior authorization from the patient’s health
insurer when necessary. Many private payors require extensive documentation of a multi-step diagnosis before authorizing SI-Joint fusion
for a patient. We believe that some private payors apply their own coverage policies and criteria inconsistently, and clinicians may
experience difficulties in securing approval and coverage for sacroiliac fusion procedures. Additionally, many private payors limit coverage
for open SI-Joint fusion to trauma, tumors or extensive spine fusion procedures involving multiple levels. The perception by physicians
that the reimbursement for SI-Joint fusion is insufficient to compensate them for the work required, including diagnosis, documentation,
obtaining payor approval for the procedure, and burden on their office staff, may negatively affect the number of procedures performed
and may therefore impede the growth of our revenues or cause them to decline.
We
may not be able to convince physicians that The Catamaran System is an attractive alternative to our competitors’ products and
that our procedure is an attractive alternative to existing surgical and non-surgical treatments of the SI-Joint.
Clinicians
play the primary role in determining the course of treatment in consultation with their patients and, ultimately, the product that will
be used to treat a patient. In order for us to sell The Catamaran System successfully, we must convince clinicians through education
and training that treatment with The Catamaran System is beneficial, safe, and cost-effective for patients as compared to our competitors’
products. If we are not successful in convincing clinicians of the merits of The Catamaran System, they may not use our product, and
we will be unable to increase our sales and achieve or grow profitability.
Historically,
most spine clinicians did not include SI-Joint pain in their diagnostic work-up because they did not have an adequate surgical procedure
to perform for patients diagnosed with the condition. As a result, some patients with lower back pain resulting from SI-Joint dysfunction
are misdiagnosed. We believe that educating clinicians and other healthcare professionals about the clinical merits and patient benefits
of The Catamaran System is an important element of our growth. If we fail to effectively educate clinicians and other medical professionals,
they may not include a SI-Joint evaluation as part of their diagnosis and, as a result, those patients may continue to receive unnecessary
or only non-surgical treatment.
Clinicians
may also hesitate to change their medical treatment practices for other reasons, including the following:
| ● | lack
of experience with minimally invasive procedures; |
| ● | perceived
liability risks generally associated with the use of new products and procedures; |
| ● | costs
associated with the purchase of new products; and |
| ● | time
commitment that may be required for training. |
Furthermore,
we believe clinicians may not widely adopt The Catamaran System unless they determine, based on experience, clinical data, and published
peer-reviewed publications, that surgical intervention provides benefits or is an attractive alternative to non-surgical treatments of
SI-Joint dysfunction. In addition, we believe support of our products relies heavily on long-term data showing the benefits of using
our product. If we are unable to provide that data, clinicians may not use our product. In such circumstances, we may not achieve expected
sales and may be unable to achieve profitability.
Clinicians
and payors may not find our clinical evidence to be compelling, which could limit our sales, and on-going and future research may prove
our product to be less safe and effective than initially anticipated.
All
of the component parts of The Catamaran System have either received premarket clearance under Section 510(k) of the U.S. federal Food,
Drug, and Cosmetic Act, or FDCA, or are exempt from premarket review. The 510(k) clearance process of the U.S. Food and Drug Administration,
or FDA, requires us to document that our product is “substantially equivalent” to another 510(k) -cleared product. The 510(k)
process is shorter and typically requires the submission of less supporting documentation than other FDA approval processes, such as
a premarket approval, or PMA, and does not usually require pre-clinical or clinical studies. Additionally, to date, we have not been
required to complete clinical studies in connection with the sale of our product. For these reasons, clinicians may be slow to adopt
our product, third-party payors may be slow to provide coverage, and we may be subject to greater regulatory and product liability risks.
Further, future patient studies or clinical experience may indicate that treatment with our product does not improve patient outcomes.
Such results would slow the adoption of our product by clinicians, significantly reduce our ability to achieve expected sales, and could
prevent us from achieving profitability. Moreover, if future results and experience indicate that our product causes unexpected or serious
complications or other unforeseen negative effects, we could be subject to mandatory product recalls, suspension, or withdrawal of FDA
clearance.
Pricing
pressure from our competitors, changes in third-party coverage and reimbursement, healthcare provider consolidation, payor consolidation
and the proliferation of “physician-owned distributorships” may impact our ability to sell our product at prices necessary
to support our current business strategies.
If
competitive forces drive down the prices we are able to charge for our product, our profit margins will shrink, which will adversely
affect our ability to invest in and grow our business. The SI-Joint fusion market has attracted numerous new companies and technologies.
As a result of this increased competition, we believe there will be continued and increased pricing pressure, resulting in lower gross
margins, with respect to our product.
Even
to the extent our product and procedures using our product are currently covered and reimbursed by third-party private and public payors,
adverse changes in coverage and reimbursement policies that affect our product, discounts, and number of implants used may also drive
our prices down and harm our ability to market and sell our product.
We
are unable to predict what changes will be made to the reimbursement methodologies used by third-party payors. We cannot be certain that
under current and future payment systems, in which healthcare providers may be reimbursed a set amount based on the type of procedure
performed, such as those utilized by Medicare and in many privately managed care systems, the cost of our product will be justified and
incorporated into the overall cost of the procedure. In addition, to the extent there is a shift from inpatient setting to outpatient
settings, we may experience pricing pressure and a reduction in the number of The Catamaran System procedures performed.
Consolidation
in the healthcare industry, including both third-party payors and healthcare providers, could lead to demands for price concessions or
to the exclusion of some suppliers from certain of our markets, which could have an adverse effect on our business, results of operations,
or financial condition. Because healthcare costs have risen significantly over the past several years, numerous initiatives and reforms
initiated by legislators, regulators, and third-party payors to curb these costs have resulted in a consolidation trend in the healthcare
industry to aggregate purchasing power. As the healthcare industry consolidates, competition to provide products and services to industry
participants has become and will continue to become more intense. This in turn has resulted and will likely continue to result in greater
pricing pressures and the exclusion of certain suppliers from important market segments as group purchasing organizations, independent
delivery networks, and large single accounts continue to use their market power to consolidate purchasing decisions for hospitals. We
expect that market demand, government regulation, third-party coverage, and reimbursement policies and societal pressures will continue
to change the worldwide healthcare industry, resulting in further business consolidations and alliances among our customers, which may
reduce competition, exert further downward pressure on the price of our product, and adversely impact our business, results of operations,
or financial condition. As we continue to expand into international markets, we will face similar risks relating to adverse changes in
coverage and reimbursement procedures and policies in those markets.
We
operate in a very competitive business environment and if we are unable to compete successfully against our existing or potential competitors,
our sales and operating results may be negatively affected and we may not grow.
The
Catamaran System is subject to intense competition. Many of our competitors are major medical device companies that have substantially
greater financial, technical, and marketing resources than we do, and they may succeed in developing products that would render our product
obsolete or non-competitive. In addition, many of these competitors have significantly longer operating histories and more established
reputations than we do. Our field is intensely competitive, subject to rapid change and highly sensitive to the introduction of new products
or other market activities of industry participants. Our ability to compete successfully will depend on our ability to develop proprietary
products that reach the market in a timely manner, receive adequate coverage and reimbursement from third-party payors, and are safer,
less invasive, and more effective than alternatives available for similar purposes as demonstrated in peer-reviewed clinical publications.
Because of the size of the potential market, we anticipate that other companies will dedicate significant resources to developing competing
products.
In
the United States, we believe that our primary competitors are currently SI-bone, Inc., Globus Medical, Inc., Medtronic plc, XTant Medical
Holdings, Inc., and RTI Surgical, Inc. At any time, these or other industry participants may develop alternative treatments, products
or procedures for the treatment of the SI-Joint that compete directly or indirectly with our product. If alternative treatments are,
or are perceived to be, superior to our product, sales of our product and our results of operations could be negatively affected. Some
of our larger competitors are either publicly traded or divisions or subsidiaries of publicly traded companies. These competitors may
enjoy several competitive advantages over us, including:
| ● | greater
financial, human, and other resources for product research and development, sales and marketing, and legal matters; |
| ● | significantly
greater name recognition; |
| ● | established
relationships with clinicians, hospitals, and other healthcare providers; |
| ● | large
and established sales and marketing and distribution networks; |
| ● | greater
experience in obtaining and maintaining domestic and international regulatory clearances or approvals, or CE Certificates of Conformity
for products and product enhancements; |
| ● | more
expansive portfolios of intellectual property rights; and |
| ● | greater
ability to cross-sell their products or to incentivize hospitals or clinicians to use their products. |
New
participants have increasingly entered the medical device industry. Many of these new competitors specialize in a specific product or
focus on a particular market segment, making it more difficult for us to increase our overall market position. The frequent introduction
by competitors of products that are or claim to be superior to our product or that are alternatives to our existing or planned products
may make it difficult to differentiate the benefits of our product over competing products. In addition, the entry of multiple new products
and competitors may lead some of our competitors to employ pricing strategies that could adversely affect the pricing of our product
and pricing in the market generally.
As
a result, without the timely introduction of new products and enhancements, our product may become obsolete over time. If we are unable
to develop innovative new products, maintain competitive pricing, and offer products that clinicians and other physicians perceive to
be as reliable as those of our competitors, our sales or margins could decrease, thereby harming our business.
We
currently manufacture (through third parties) and sell products used in a single procedure, which could negatively affect our operations
and financial condition.
Presently
we do not sell any products other than The Catamaran System and related tools and instruments. Therefore, we are solely dependent on
widespread market adoption of The Catamaran System and we will continue to be dependent on the success of this single product for the
foreseeable future. There can be no assurance that The Catamaran System will gain a substantial degree of market acceptance among clinicians,
patients or healthcare providers. Our failure to successfully increase sales of The Catamaran System or any other event impeding our
ability to sell The Catamaran System would result in a material adverse effect on our results of operations, financial condition and
continuing operations.
We
have a limited operating history and may face difficulties encountered by early-stage companies in new and rapidly evolving markets.
Even
though we were formed in 2012 we have just built the infrastructure necessary to commercially launch The Catamaran System. Accordingly,
we have a limited operating history upon which to base an evaluation of our business and prospects. In assessing our prospects, you must
consider the risks and difficulties frequently encountered by early-stage companies in new and rapidly evolving markets, particularly
companies engaged in the development and sales of medical devices. These risks include our inability to:
| ● | obtain
coverage by third-party, private, and government payors; |
| ● | establish
and increase awareness of our brand and strengthen customer loyalty; |
| ● | attract
and retain qualified personnel; |
| ● | find
and develop relationships with contract manufacturers that can manufacture the necessary volume of product; |
| ● | manage
our independent sales representatives to achieve our sales growth objectives; |
| ● | commercialize
new products and enhance our existing product; |
| ● | manage
rapidly changing and expanding operations; |
| ● | implement
and successfully execute our business and marketing strategy; |
| ● | respond
effectively to competitive pressures and developments. |
We
can also be negatively affected by general economic conditions. Because of our limited operating history, we may not have insight into
trends that could emerge and negatively affect our business. As a result of these or other risks, our business strategy might not be
successful.
Our
sales volumes and our operating results may fluctuate over the course of the year.
Since
we had our first sales in April 2021 and our official national launch commenced in October 2022, we have limited history with respect
to how rapidly adoption of The Catamaran System will occur. Sales growth could be slower than we have projected. Our sales and results
of operations will be affected by numerous factors, including, among other things:
| ● | payor
coverage and reimbursement; |
| ● | maintaining
our training schedule with clinicians; |
| ● | the
number of procedures performed in the quarter and our ability to drive increased sales of our product; |
| ● | our
ability to identify and sign-up independent sales representatives and their performance; |
| ● | pricing
pressure applicable to our product, including adverse third-party coverage and reimbursement outcomes; |
| ● | timing
of new product offerings, acquisitions, licenses or other significant events by us or our competitors; |
| ● | our
ability to find and develop relationships with contract manufacturers and their ability to timely provide us with an adequate supply
of products; |
| ● | the
evolving product offerings of our competitors; |
| ● | the
demand for, and pricing of, our product and the products of our competitors; |
| ● | factors
that may affect the sale of our product, including seasonality and budgets of our customers; |
| ● | interruption
in the manufacturing or distribution of our product; |
| ● | the
effect of competing technological, industry and market developments; |
| ● | our
ability to expand the geographic reach of our sales and marketing efforts; |
| ● | the
costs of maintaining adequate insurance coverage, including product liability insurance; |
| ● | the
availability and cost of components and materials needed by our contract manufacturers; |
| ● | the
number of selling days in the quarter; and |
| ● | impairment
and other special charges. |
Some
of the products we may seek to develop and introduce in the future will require FDA clearance or approval before commercialization in
the United States. As a result, it will be difficult for us to forecast demand for these products with any degree of certainty. In addition,
we will be increasing our operating expenses as we expand our commercial capabilities. Accordingly, we may experience significant, unanticipated
quarterly losses. If our quarterly or annual operating results fall below the expectations of investors or securities analysts, the price
of our common stock could decline substantially. Furthermore, any quarterly or annual fluctuations in our operating results may, in turn,
cause the price of our common stock to fluctuate substantially. Quarterly comparisons of our financial results may not always be meaningful
and should not be relied upon as an indication of our future performance.
If
we do not successfully implement our business strategy, our business and results of operations will be adversely affected.
Our
business strategy was based on assumptions about the market that might prove wrong. We believe that various demographics and industry-specific
trends will help drive growth in the market and our business, but these demographics and trends have been and will continue to be uncertain.
Actual demand for our product could differ materially from projected demand if our assumptions regarding these factors prove to be incorrect
or do not materialize, or if alternative treatments to those offered by our product gains widespread acceptance. Also, our strategy of
focusing exclusively on the SI-Joint market may limit our ability to grow. In addition, in order to increase our sales, we will need
to identify and contract with independent sales representatives in existing and new regions as well, and in the future, commercialize
new products. Moreover, we may decide to alter or discontinue aspects of our business strategy and may adopt different strategies due
to business or competitive factors not currently foreseen, such as new medical technologies that would make our product obsolete. Any
failure to implement our business strategy may adversely affect our business, results of operations, and financial condition.
Our
business could suffer if we lose the services of key members of our senior management, key advisors or personnel.
We
are dependent upon the continued services of key members of our senior management and a number of key advisors and personnel. The loss
of members of our senior management team, key advisors or personnel, or our inability to attract or retain other qualified personnel
or advisors, could have a material adverse effect on our business, results of operations, and financial condition. We do not maintain
“key person” insurance for any of our executives or employees. In addition, several of the members of our executive management
team are not subject to non-competition agreements that restrict their ability to compete with us. Accordingly, the adverse effect resulting
from the loss of certain executives could be compounded by our inability to prevent them from competing with us.
Various
factors outside our direct control may adversely affect manufacturing and distribution of our product.
The
manufacture and distribution of our product is challenging. Changes that our contract manufacturers may make outside the purview of our
direct control can have an impact on our processes, quality of our product, and the successful delivery of products to our customers.
Mistakes and mishandling are not uncommon and can affect supply and delivery. Some of these risks include:
| ● | failure
to manufacture in compliance with the required regulatory standards; |
| ● | the
cost and availability of components and supplies required by our contract manufacturers to manufacture our products; |
| ● | delays
in analytical results or failure of analytical techniques that we will depend on for quality control and release of products; |
| ● | natural
disasters, labor disputes, financial distress, raw material availability, issues with facilities and equipment, or other forms of disruption
to business operations affecting our manufacturers or their suppliers; and |
| ● | latent
defects that may become apparent after products have been released and that may result in a recall of such products. |
If
any of these risks were to materialize, our ability to provide our product to customers on a timely basis would be adversely impacted.
We
are dependent on a limited number of contract manufacturers, some of them single-source and some of them in single locations, for our
product, and the loss of any of these contract manufacturers, or their inability to provide us with an adequate supply of products in
a timely and cost-effective manner, could materially adversely affect our business.
We
rely on contract manufacturers to supply our product. For us to be successful, our contract manufacturers must be able to provide us
with product in substantial quantities, in compliance with regulatory requirements, in accordance with agreed upon specifications, at
acceptable prices, and on a timely basis. We have a limited history with our current contract manufacturers and do not have long-term
supply contracts with them. We are in the process of identifying and evaluating new contract manufacturers for our product. The inability
to find the required contract manufacturers or the time required to switch contract manufacturers could adversely affect sales.
In
addition, our anticipated growth could strain the ability of our contract manufacturers to deliver an increasingly large supply of product.
Contract manufacturers often experience difficulties in scaling up production, including financial issues, or problems with production
yields and quality control and assurance.
We
use a small number of contract manufacturers for our instruments. Our dependence on such a limited number of contract manufacturers exposes
us to risks, including, among other things:
| ● | contract
manufacturers may fail to comply with regulatory requirements or make errors in manufacturing that could negatively affect the safety
or effectiveness of our product or cause delays in shipments of our product; |
| ● | some
of our contract manufacturers have long lead times of 12 to 16 weeks and we may not be able to respond to unanticipated changes in customer
orders, and if orders do not match forecasts, we or our contract manufacturers may have excess or inadequate inventory of materials and
components; |
| ● | our
contract manufacturers may be subject to price fluctuations due to a lack of long-term supply arrangements for key components; |
| ● | our
contract manufacturers may lose access to critical services and components, resulting in an interruption in the manufacture, assembly
and shipment of our product; |
| ● | we
may experience delays in delivery by our contract manufacturers due to changes in demand from us or their other customers; |
| ● | fluctuations
in demand for products that our contract manufacturers manufacture for others may affect their ability or willingness to deliver our
product to us in a timely manner; |
| ● | our
contract manufacturers may wish to discontinue supplying products or services to us for risk management reasons; |
| ● | we
may not be able to find new or alternative contract manufacturers in a timely manner if our current contract manufacturers stop producing
products; and |
| ● | our
contract manufacturers may encounter financial hardships unrelated to our demand, which could inhibit their ability to fulfil our orders
and meet our requirements. |
If
any one or more of these risks materialize, it could significantly increase our costs and impact our ability to meet demand for our product.
If we are unable to satisfy commercial demand for our product in a timely manner, our ability to generate revenue would be impaired,
market acceptance of our product could be adversely affected, and customers may instead purchase or use our competitors’ products.
Additionally, we could be forced to seek alternative sources of supply.
Because
of the nature of our internal quality control requirements, regulatory requirements, and the custom and proprietary nature of our product,
we may not be able to quickly engage additional or replacement contract manufacturers for our product and accessories. We may also be
required to assess any potential new contract manufacturer’s compliance with all applicable regulations and guidelines, which could
further impede our ability to obtain our product in a timely manner. As a result, we could incur increased product costs, experience
delays in deliveries of our product, suffer damage to our reputation, and experience an adverse effect on our business and financial
results. Failure of any of our contract manufacturers to meet our product demand level would limit our ability to meet our sales commitments
to our customers and could have a material adverse effect on our business.
We
may also have difficulty obtaining similar product from other contract manufacturers that are acceptable to the FDA and the failure of
our contract manufacturers to comply with strictly enforced regulatory requirements could expose us to delays in obtaining clearances
or approvals, regulatory action including warning letters, product recalls, termination of distribution, product seizures, civil, administrative,
or criminal penalties. We could incur delays while we locate and engage qualified alternative contract manufacturers, and we may be unable
to engage alternative contract manufacturers on favorable terms or at all. Any such disruption or increased expenses could harm our commercialization
efforts and adversely affect our ability to generate sales.
In
addition, we expect that most of our contract manufacturers will operate at a facility in a single location and substantially all their
inventory of component supplies and finished goods will be held at these locations. We, and our contract manufacturers, will take precautions
to safeguard facilities, including acquiring insurance, adopting health and safety protocols, and utilizing off-site storage of computer
data. However, vandalism, terrorism, or a natural or other disaster, such as an earthquake, fire, or flood, could damage or destroy equipment
or component supplies or finished product, cause substantial delays in our operations, result in the loss of key information, and cause
us to incur additional expenses. Our insurance may not cover our losses in any particular case. In addition, regardless of the level
of insurance coverage, damage to our or our contract manufacturers’ facilities could harm our business, financial condition, and
operating results.
As
our sales grow, our contract manufacturers may encounter problems or delays in the manufacturing of our product or fail to meet certain
regulatory requirements which could result in an adverse effect on our business and financial results.
To
become profitable, our contract manufactures must manufacture our product in adequate quantities in compliance with regulatory requirements
and at an acceptable cost. Increasing their capacity to manufacture and inspect our product may require them to improve internal efficiencies
or require us to re-design or change the specifications of our product. Our contract manufacturers may encounter several difficulties
in increasing this capacity, including:
| ● | managing
production yields; |
| ● | maintaining
quality control and assurance; |
| ● | providing
component and service availability; |
| ● | maintaining
adequate control policies and procedures; |
| ● | hiring
and retaining qualified personnel; and |
| ● | complying
with state, federal, and foreign regulations. |
If
we are unable to satisfy commercial demand for The Catamaran System due to our contract manufacturer’s inability to manufacture
and inspect our product, our ability to generate revenue would be impaired, market acceptance of our product could be adversely affected
and customers may instead purchase or use our competitors’ products.
The
size and future growth in the market for the SI-Joint fixation market have not been established based on market reports and our estimates
are based on our own review and analysis of public information and may be smaller than we estimate, possibly materially. In addition,
our estimates of cost savings to the economy and healthcare system as a result of The Catamaran System procedure are based on our internal
estimates and market research and could also be smaller than we estimate, possibly materially. If our estimates and projections overestimate
the size of this market or cost savings, our sales growth may be adversely affected.
We
are not aware of an independent third-party study that reliably reports the potential market size for the SI-Joint fixation market. Therefore,
our estimates of the size and future growth in the market for The Catamaran System product, including cost savings to the economy overall,
including patients and employers, and to the healthcare system and the number of people currently suffering from lower back pain who
may benefit from and be amenable to our procedure, is based on a number of internal and third-party studies, surveys, reports, and estimates.
While we believe these factors have historically provided and may continue to provide us with effective tools in estimating the total
market for our product and procedures and health cost savings, these estimates may not be correct and the conditions supporting our estimates
may change at any time, thereby reducing the predictive accuracy of these underlying factors. For example, we have consulted with our
clinical advisors and utilized public information as the basis for our market projections. Additionally, the surveys we have conducted
are based on a small number of respondents and are not statistically significant and may have other limitations. The actual incidence
of lower back pain, and the actual demand for our product or competitive products, could differ materially from our projections if our
assumptions and estimates are incorrect. As a result, our estimates of the size and future growth in the market for our product may prove
to be incorrect. In addition, actual health cost savings to the healthcare system as a result of The Catamaran System procedure may materially
differ from those presented in this report. If the actual number of people with lower back pain who would benefit from The Catamaran
System and the size and future growth in the market and related costs savings to the healthcare system is smaller than we have estimated,
it may impair our projected sales growth and have an adverse impact on our business.
In
the future our product may become obsolete, which would negatively affect operations and financial condition.
The
medical device industry is characterized by rapid and significant change. There can be no assurance that other companies will not succeed
in developing or marketing devices, and products that are more effective than The Catamaran System or that would render The Catamaran
System obsolete or non-competitive. Additionally, new surgical procedures, medications and other therapies could be developed that replace
or reduce the importance of our product. Accordingly, our success will depend in part on our ability to respond quickly to medical and
changes through the development and introduction of new products. Product development involves a high degree of risk and there can be
no assurance that our new product development efforts will result in any commercially successful products.
If
we experience significant disruptions in our information technology systems, our business, results of operations, and financial
condition could be adversely affected.
The
efficient operation of our business depends on our information technology systems. We will rely on our information technology systems
to effectively manage:
| ● | sales
and marketing, accounting, and financial functions; |
| ● | engineering
and product development tasks; and |
| ● | our
research and development data. |
Our
information technology systems are vulnerable to damage or interruption from:
| ● | earthquakes,
fires, floods, and other natural disasters; |
| ● | terrorist
attacks and attacks by computer viruses or hackers; |
| ● | computer
systems, or Internet, telecommunications, or data network failures. |
The
failure of our information technology systems to perform as we anticipate or our failure to effectively implement new systems could disrupt
our entire operation and could result in decreased sales, increased overhead costs, excess inventory and product shortages, and legal
liability issues, all of which could have a material adverse effect on our reputation, business, results of operations, and financial
condition.
We
may seek to grow our business through acquisitions of or investments in new or complementary businesses, products or technologies, and
the failure to manage acquisitions or investments, or the failure to integrate them with our existing business, could have a material
adverse effect on us.
From
time to time, we expect to consider opportunities to acquire or make investments in other technologies, products, and businesses that
may enhance our capabilities, complement our current product, or expand the breadth of our markets or customer base. Potential and completed
acquisitions and strategic investments involve numerous risks, including:
| ● | problems
assimilating the purchased technologies, products, or business operations; |
| ● | issues
maintaining uniform standards, procedures, controls, and policies; |
| ● | unanticipated
costs and liabilities associated with acquisitions; |
| ● | diversion
of management’s attention from our core business; |
| ● | adverse
effects on existing business relationships with suppliers and customers; |
| ● | risks
associated with entering new markets in which we have limited or no experience; |
| ● | potential
loss of key employees of acquired businesses; and |
| ● | increased
legal and accounting compliance costs. |
We
have no current commitments with respect to any acquisition or investment. We do not know if we will be able to identify acquisitions,
we deem suitable, whether we will be able to successfully complete any such acquisitions on favorable terms or at all, or whether we
will be able to successfully integrate any acquired business, product, or technology into our business or retain any key personnel, suppliers,
or distributors. Our ability to successfully grow through acquisitions depends upon our ability to identify, negotiate, complete, and
integrate suitable target businesses and to obtain any necessary financing. These efforts could be expensive and time consuming and may
disrupt our ongoing business and prevent management from focusing on our operations. If we are unable to successfully integrate any acquired
businesses, products, or technologies effectively, our business, results of operations, and financial condition will be materially adversely
affected.
We
may enter into collaborations, in-licensing arrangements, joint ventures, strategic alliances, or partnerships with third-parties that
may not result in the development of commercially viable products or the generation of significant future revenue.
In
the ordinary course of our business, we may enter into collaborations, in-licensing arrangements, joint ventures, strategic alliances,
partnerships, or other arrangements to develop products and to pursue new markets. We have not entered into any collaboration arrangements
to date. Proposing, negotiating, and implementing collaborations, in-licensing arrangements, joint ventures, strategic alliances, or
partnerships may be a lengthy and complex process. Other companies, including those with substantially greater financial, marketing,
sales, technology, or other business resources, may compete with us for these opportunities or arrangements. We may not identify, secure,
or complete any such transactions or arrangements in a timely manner, on a cost-effective basis, on acceptable terms or at all. We have
limited institutional knowledge and experience with respect to these business development activities, and we may also not realize the
anticipated benefits of any such transaction or arrangement. These collaborations may not result in the development of products that
achieve commercial success or result in significant revenue and could be terminated prior to developing any products.
Additionally,
we may not be able to exercise sole decision-making authority regarding the transaction or arrangement, which could create the potential
risk of creating impasses on decisions, and our future collaborators may have economic or business interests or goals that are, or that
may become, inconsistent with our business interests or goals. It is possible that conflicts may arise with our collaborators, such as
conflicts concerning the achievement of performance milestones, or the interpretation of significant terms under any agreement, such
as those related to financial obligations or the ownership or control of intellectual property developed during the collaboration. If
any conflicts arise with any future collaborators, they may act in their self- interest, which may be adverse to our best interest, and
they may breach their obligations to us. In addition, we may have limited control over the amount and timing of resources that any future
collaborators devote to our or their future products.
Disputes
between us and our collaborators may result in litigation or arbitration which would increase our expenses and divert the attention of
our management. Further, these transactions and arrangements will be contractual in nature and will generally be terminable under the
terms of the applicable agreements and, in such event, we may not continue to have rights to the products relating to such transaction
or arrangement or may need to purchase such rights at a premium. If we enter into in-bound intellectual property license agreements,
we may not be able to fully protect the licensed intellectual property rights or maintain those licenses. Future licensors could retain
the right to prosecute and defend the intellectual property rights licensed to us, in which case we would depend on the ability of our
licensors to obtain, maintain and enforce intellectual property protection for the licensed intellectual property. These licensors may
determine not to pursue litigation against other companies or may pursue such litigation less aggressively than we would. Further, entering
into such license agreements could impose various diligence, commercialization, royalty, or other obligations on us. Future licensors
may allege that we have breached our license agreement with them, and accordingly seek to terminate our license, which could adversely
affect our competitive business position and harm our business prospects.
We
are increasingly dependent on information technology, and our systems and infrastructure face certain risks, including cybersecurity
and data leakage risks.
Significant
disruptions to our information technology systems or breaches of information security could adversely affect our business. In the ordinary
course of business, we will collect, store and transmit large amounts of confidential information, and it is critical that we do so in
a secure manner to maintain the confidentiality and integrity of such information. We have also outsourced significant elements of our
information technology infrastructure; as a result, we manage independent vendor relationships with third parties who are responsible
for maintaining significant elements of our information technology systems and infrastructure and who may or could have access to our
confidential information. The size and complexity of our information technology systems, and those of our third-party vendors, make such
systems potentially vulnerable to service interruptions and security breaches from inadvertent or intentional actions by our employees,
partners or vendors. These systems are also vulnerable to attacks by malicious third parties and may be susceptible to intentional or
accidental physical damage to the infrastructure maintained by us or by third parties. Maintaining the secrecy of confidential, proprietary
and/or trade secret information is important to our competitive business position. While we have taken steps to protect such information
and have invested in systems and infrastructures to do so, there can be no guarantee that our efforts will prevent service interruptions
or security breaches in our systems or the unauthorized or inadvertent wrongful use or disclosure of confidential information that could
adversely affect our business operations or result in the loss, dissemination or misuse of critical or sensitive information. The increasing
sophistication and frequency of cybersecurity threats, including targeted data breaches, ransomware attacks designed to encrypt our data
for ransom and other malicious cyber activities, pose a significant risk to the integrity and confidentiality of our data systems. A
breach our security measures or the accidental loss, inadvertent disclosure, unapproved dissemination, misappropriation or misuse of
trade secrets, proprietary information or other confidential information, whether as a result of theft, hacking, fraud, trickery or other
forms of deception, or for any other cause, could enable others to produce competing products, use our proprietary technology or information,
and/or adversely affect our business position. Further, any such interruption, security breach, loss or disclosure of confidential information
could result in financial, legal, business and reputational harm to us and could have a material adverse effect on our business, financial
position, results of operations and/or cash flow.
Geopolitical
conditions, including trade disputes and direct or indirect acts of war or terrorism, could have an adverse effect on our operations
and financial results.
Our
operations could be disrupted by geopolitical conditions, political and social instability, acts of war, terrorist activity or other
similar events. In February 2022, Russia initiated significant military action against Ukraine. In response, the U.S. and certain other
countries imposed significant sanctions and export controls against Russia, Belarus and certain individuals and entities connected to
Russian or Belarusian political, business, and financial organizations, and the U.S. and certain other countries could impose further
sanctions, trade restrictions, and other retaliatory actions should the conflict continue or worsen. It is not possible to predict the
broader consequences of the conflict, including related geopolitical tensions, and the measures and retaliatory actions taken by the
U.S. and other countries in respect thereof as well as any counter measures or retaliatory actions by Russia or Belarus in response,
including, for example, potential cyberattacks or the disruption of energy exports, is likely to cause regional instability, geopolitical
shifts, and could materially adversely affect global trade, currency exchange rates, regional economies and the global economy. In addition,
the ongoing conflicts in the Middle East may further impact global economic conditions and market sentiments. This, in turn, could adversely
affect the trading price of our shares of common stock and investor interest in us. The outcome of the Russia-Ukraine war and conflicts
in the Middle East remain uncertain, and while it is difficult to predict the impact of any of the foregoing, the conflict and actions
taken in response to the conflict could increase our costs, disrupt our supply chain, reduce our sales and earnings, impair our ability
to raise additional capital when needed on acceptable terms, if at all, or otherwise adversely affect our business, financial condition,
and results of operations.
Inflation
may adversely affect our operations and financial results.
In
periods of rising inflation, the cost of raw materials, components and labor essential for manufacturing The Catamaran System may increase
and as a consequence, our overall profit margin may be adversely affected. In addition, inflation may result in limitations on healthcare
spending, specifically for procedures that are deemed elective or non-critical, which may include treatments utilizing The Catamaran
System. A decrease in demand for these procedures may significantly impact our financial condition and results of operations.
Risks
Related to Our Legal and Regulatory Environment
We
and our contract manufacturers are subject to extensive governmental regulation both in the United States and abroad, and failure to
comply with applicable requirements could cause our business to suffer.
The
medical device industry is regulated extensively by governmental authorities, principally the FDA and corresponding state and foreign
regulatory agencies. The FDA and other U.S. and foreign governmental agencies regulate, among other things, with respect to medical devices:
| ● | design,
development, and manufacturing; |
| ● | testing,
labeling, content, and language of instructions for use and storage; |
| ● | marketing,
sales, and distribution; |
| ● | premarket
clearance and approval; |
| ● | conformity
assessment procedures; |
| ● | record
keeping procedures; |
| ● | advertising
and promotion; |
| ● | compliance
with good manufacturing practices requirements; |
| ● | recalls
and field safety corrective actions; |
| ● | post-market
surveillance, including reporting of deaths or serious injuries and malfunctions that, if they were to recur, could lead to death or
serious injury; |
| ● | post-market
approval studies; and |
| ● | product
import and export. |
The
regulations to which we are subject are complex and have tended to become more stringent over time. Regulatory changes could result in
restrictions on our ability to carry on or expand our operations, difficulties achieving new product clearances, higher than anticipated
costs or lower than anticipated sales.
Before
we can market or sell a new regulated product or make a significant modification to an existing product in the United States, with very
limited exception, we must obtain either clearance under Section 510(k) of the FDCA for Class II devices or approval of a premarket approval
application from the FDA for a Class III device. In the 510(k) clearance process, the FDA must determine that a proposed device is “substantially
equivalent” to a device legally on the market, known as a “predicate” device, with respect to intended use, technology,
and safety and effectiveness, in order to clear the proposed device for marketing. Clinical data is sometimes required to support substantial
equivalence. The PMA pathway requires an applicant to demonstrate the safety and effectiveness of the device based, in part, on extensive
data, including, but not limited to, technical, preclinical, clinical trial, manufacturing, and labeling data. The PMA process is typically
required for devices that are deemed to pose the greatest risk, such as life-sustaining, life-supporting, or implantable devices. Products
that are approved through a PMA application generally need FDA approval before they can be modified. Similarly, some modifications made
to products cleared through a 510(k) may require a new 510(k). Both the 510(k) and PMA processes can be expensive and lengthy and require
the payment of significant fees, unless exempt. The FDA’s 510(k) clearance process usually takes from three to 12 months but may
last longer. The process of obtaining a PMA is much more costly and uncertain than the 510(k) clearance process and generally takes from
one to three years, or even longer, from the time the application is submitted to the FDA until an approval is obtained. The process
of obtaining domestic and international regulatory clearances or approvals to market a medical device can be costly and time consuming,
and we may not be able to obtain these clearances or approvals on a timely basis, if at all.
In
the United States, all of the components to The Catamaran System have either received premarket clearance under Section 510(k) of the
FDCA or are exempt from premarket review. If the FDA requires us to go through a lengthier, more rigorous examination for future products
or modifications to existing products than we had expected, our product introductions or modifications could be delayed or canceled,
which could cause our sales to decline. In addition, the FDA may determine that future products will require the more costly, lengthy,
and uncertain PMA process. Although we do not currently market any devices under PMA, the FDA may demand that we obtain a PMA prior to
marketing certain of our future products. In addition, if the FDA disagrees with our determination that a product, we currently market
is subject to an exemption from premarket review, the FDA may require us to submit a 510(k) or PMA in order to continue marketing the
product. Further, even with respect to those future products where a PMA is not required, we cannot assure you that we will be able to
obtain the 510(k) clearances with respect to those products.
The
FDA can delay, limit or deny clearance or approval of a device for many reasons, including:
| ● | we
may not be able to demonstrate to the FDA’s satisfaction that our product is safe and effective for their intended users; |
| ● | the
data from our pre-clinical studies and clinical trials may be insufficient to support clearance or approval, where required; and |
| ● | the
manufacturing process or facilities we use may not meet applicable requirements. |
In
addition, the FDA may change its clearance and approval policies, adopt additional regulations or revise existing regulations, or take
other actions which may prevent or delay clearance or approval of our product under development or impact our ability to modify our currently
approved or cleared product on a timely basis.
Any
delay in, or failure to receive or maintain, clearance or approval for our product under development could prevent us from generating
revenue from these products or achieving profitability.
In
addition, even after we have obtained the proper regulatory clearance or approval to market a product, the FDA has the power to require
us to conduct post-market surveillance on our product. These studies can be very expensive and time consuming to conduct. Failure to
comply with those studies in a timely manner could result in the revocation of the 510(k) clearance for a product that is subject to
such surveillance and the recall or withdrawal of the product, which could prevent us from generating sales from that product in the
United States.
Additionally,
as part of the conformity assessment process, medical device manufacturers must carry out a clinical evaluation of their medical devices
to verify that they comply with the relevant Essential Requirements covering safety and performance. A clinical evaluation includes an
assessment of whether a medical device’s performance is in accordance with its intended use and that the known and foreseeable
risks linked to the use of the device under normal conditions are minimized and acceptable when weighed against the benefits of its intended
purpose. The clinical evaluation conducted by the manufacturer must also address any clinical claims, the adequacy of the device labeling
and information (particularly claims, contraindications, precautions/ warnings) and the suitability of related Instructions for Use.
This assessment must be based on clinical data, which can be obtained from (i) clinical studies conducted on the devices being assessed;
(ii) scientific literature from similar devices whose equivalence with the assessed device can be demonstrated; or (iii) both clinical
studies and scientific literature.
The
FDA and other regulatory authorities have broad enforcement powers. Regulatory enforcement or inquiries, or other increased scrutiny
on us, could dissuade some clinicians from using our product and adversely affect our reputation and the perceived safety and effectiveness
of our product.
Failure
to comply with applicable regulations could jeopardize our ability to sell our product and result in enforcement actions such as:
| ● | termination
of distribution; |
| ● | recalls
or seizures of products; |
| ● | delays
in the introduction of products into the market; |
| ● | total
or partial suspension of production; |
| ● | refusal
of the FDA other regulators to grant future clearances or approvals; or |
| ● | in
the most serious cases, criminal penalties. |
Adverse
action by an applicable regulatory agency the FDA could result in inability to produce our product in a cost-effective and timely manner,
or at all, decreased sales, higher prices, lower margins, additional unplanned costs or actions, damage to our reputation, and could
have material adverse effect on our reputation, business, results of operations, and financial condition.
We
and our independent sales representatives must comply with U.S. federal and state fraud and abuse laws, including those relating to physician
kickbacks and false claims for reimbursement.
Healthcare
providers, distributors, physicians, and third-party payors play a primary role in the distribution, recommendation, ordering, and purchasing
of any implant or other medical device for which we have or obtain marketing clearance or approval. Through our arrangements with customers
and third-party payors, we are exposed to the risk that our employees, independent contractors, principal investigators, consultants,
vendors, or independent sales representatives may engage in fraudulent or other illegal activity. Misconduct by these parties could include,
among other infractions or violations, intentional, reckless and/or negligent conduct or unauthorized activity that violates FDA regulations,
manufacturing standards, federal and state healthcare fraud and abuse laws and regulations, laws that require the true, complete, and
accurate reporting of financial information or data, other commercial or regulatory laws or requirements, and equivalent foreign rules.
We plan to implement a compliance program, code of conduct, and associated policies and procedures, but it is not always possible to
identify and deter misconduct by our employees and other third parties, and the precautions we plan to take to detect and prevent this
activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations
or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations, and government authorities may
conclude that our business practices do not comply with applicable fraud and abuse or other healthcare laws and regulations or guidance
despite our good faith efforts to comply.
There
are numerous U.S. federal and state laws pertaining to healthcare fraud and abuse, including anti-kickback and false claims laws. Our
relationships with clinicians, other healthcare professionals, and hospitals are subject to scrutiny under these laws.
Healthcare
fraud and abuse laws and related regulations are complex, and even minor irregularities can potentially give rise to claims that a statute
or prohibition has been violated. The laws that may affect our ability to operate include:
| ● | the
federal Anti-Kickback Statute, which prohibits, among other things, knowingly and willfully soliciting, receiving, offering or paying
remuneration, directly or indirectly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase,
order or recommendation of, items or services for which payment may be made, in whole or in part, under federal healthcare programs,
such as the Medicare and Medicaid programs; |
| ● | the
federal False Claims Act, which prohibits, among other things, individuals or entities from knowingly presenting, or causing to be presented,
false or fraudulent claims for payment of government funds; knowingly making, using, or causing to be made or used, a false record or
statement to get a false claim paid or to avoid, decrease, or conceal an obligation to pay money to the federal government. A claim including
items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes
of the False Claims Act. There are also criminal penalties for making or presenting a false or fictitious or fraudulent claim to the
federal government; |
| ● | the
federal Health Insurance Portability and Accountability Act of 1996, which imposes criminal and civil liability for, among other actions,
knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program including private third-party
payors, or knowingly and willfully falsifying, concealing, or covering up a material fact or making a materially false, fictitious, or
fraudulent statement or representation, or making or using any false writing or document knowing the same to contain any materially false,
fictitious, or fraudulent statement or entry in connection with the delivery of or payment for healthcare benefits, items, or services; |
| ● | the
federal Physician Payment Sunshine Act, which requires manufacturers of drugs, devices, biologics and medical supplies for which payment
is available under Medicare, Medicaid, or the Children’s Health Insurance Program to report annually to the Centers for Medicare
& Medicaid Services information related to payments or other “transfers of value” made to physicians and teaching hospitals,
and requires applicable manufacturers to report annually to CMS ownership and investment interests held by physicians and their immediate
family members and payments or other “transfers of value” to such physician owners; and |
| ● | analogous
state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws, which may apply to items or services
reimbursed by any third-party payor, including commercial insurers; state laws that require device companies to comply with the industry’s
voluntary compliance guidelines and the applicable compliance guidance promulgated by the federal government or otherwise restrict payments
that may be made to healthcare providers and other potential referral sources; state beneficiary inducement laws, and state laws that
require device manufacturers to report information related to payments and other transfers of value to physicians and other healthcare
providers or marketing expenditures, many of which differ from each other in significant ways and may not have the same effect, thus
complicating compliance efforts. |
If
we or our employees are found to have violated any of the above laws we may be subjected to administrative, civil and criminal penalties,
including imprisonment, exclusion from participation in federal healthcare programs, such as Medicare and Medicaid, and significant fines,
monetary penalties and damages, and damage to our reputation. Additional information about these laws is provided in “Business-Regulation.”
We
have entered into consulting agreements with clinicians who are also customers. We anticipate entering into additional agreements with
clinicians who use our product as we continue to commercialize our product. The primary mission of these clinician advisors is research
and development and clinician education. Medical device technology development requires thoughtful clinician input from experienced healthcare
professionals. Medical device clinician education requires experienced faculty for didactic and anatomic lab activities in a peer-to-peer
setting. We believe these engagements will allow us to successfully meet the expectations of the physician community. In addition, a
small number of clinicians (which are or may become customers) own less than 1.0% of our stock, or were granted stock options which they
either purchased in an arm’s length transaction on terms identical to those offered to others or received from us as fair market
value consideration for consulting services performed. While all of these transactions were structured with the intention of complying
with all applicable laws, including the federal Anti-Kickback Statute, state anti-kickback laws and other applicable laws, to the extent
applicable, it is possible that regulatory agencies may view these transactions as prohibited arrangements that must be restructured,
or discontinued, or for which we could be subject to significant penalties. We would be materially and adversely affected if regulatory
agencies interpret our financial relationships with clinicians who order our product to be in violation of applicable laws and we were
unable to comply with such laws, which could subject us to, among other things, monetary penalties for non-compliance, the cost of which
could be substantial.
In
certain cases, federal and state authorities pursue actions for false claims on the basis that manufacturers and distributors are promoting
unapproved, or “off-label” uses of their products. Pursuant to FDA regulations, we can only market our product for cleared
or approved uses. Although clinicians are permitted to use medical devices for indications other than those cleared or approved by the
FDA, we are prohibited from promoting products for “off-label” uses. We market our product and provide promotional materials
and training programs to clinicians regarding the use of our product. If it is determined that our marketing, promotional materials or
training programs constitute promotion of unapproved uses, we could be subject to significant fines in addition to regulatory enforcement
actions, including the issuance of a warning letter, injunction, seizure, criminal penalty, and damage to our reputation. Federal and
state authorities also pursue actions for false claims based upon improper billing and coding advice or recommendations, as well as decisions
related to the medical necessity of procedures, including the site-of-service where procedures are performed. Actions under the federal
False Claims Act may also be brought by whistleblowers under its qui tam provisions.
To
enforce compliance with the federal laws, the U.S. Department of Justice has increased its scrutiny of interactions between healthcare
companies and healthcare providers, which has led to a number of investigations, prosecutions, convictions and settlements in the healthcare
industry. Dealing with investigations can be time and resource consuming and can divert management’s attention from the business.
Additionally, if a healthcare company settles an investigation with the Department of Justice or other law enforcement agencies, it may
need to agree to additional onerous compliance and reporting requirements as part of a consent decree or corporate integrity agreement.
Any such investigation or settlement could increase our costs or otherwise have an adverse effect on our business. Even if we are not
determined to have violated these laws, government investigations into these issues typically require the expenditure of significant
resources and generate negative publicity, which could harm our financial condition and divert resources and the attention of our management
from operating our business.
The
scope and enforcement of these laws is uncertain and subject to rapid change. The shifting compliance environment and the need to build
and maintain robust and expandable systems to comply with different compliance and/or reporting requirements in multiple jurisdictions
increase the possibility that we may run afoul of one or more of the requirements or that federal or state regulatory authorities might
challenge our current or future activities under these laws. Additionally, we cannot predict the impact of any changes in these laws,
whether or not retroactive.
Our
failure to adequately protect personal information in compliance with evolving legal requirements could harm our business.
In
the ordinary course of our business, we plan to collect and store sensitive data, including legally protected personally identifiable
information. We may collect this kind of information during the course of future clinical trials and for possible post-marketing safety
vigilance, helping enable clinicians and their patients to pursue claims for reimbursement for procedures using The Catamaran System
and servicing potential warranty claims.
There
are a number of state, federal, and international laws protecting the privacy and security of health information and personal data. These
data protection and privacy-related laws and regulations are evolving and may result in ever-increasing regulatory and public scrutiny
of companies’ data practices and escalating levels of enforcement and sanctions. As part of the American Recovery and Reinvestment
Act 2009, or ARRA, Congress amended the privacy and security provisions of the Health Insurance Portability and Accountability Act, or
HIPAA. HIPAA imposes certain requirements regarding the privacy, security, use, and disclosure of an individual’s protected health
information, or PHI, by certain health care providers, health care clearinghouses, and health insurance plans, collectively referred
to as “covered entities,” and their “business associates,” or subcontractors who provide services to covered
entities that involve the creation, use, maintenance, or disclosure of PHI. ARRA included significant increases in the penalties for
improper use or disclosure of an individual’s PHI under HIPAA and extended enforcement authority to state attorneys general. The
amendments also created notification requirements applicable to covered entities and business associates in certain cases when PHI in
their control has been inappropriately accessed or disclosed. In the case of a breach of unsecured PHI, covered entities may be required
to provide notification to individuals affected by the breach, federal regulators, and, in some cases, local and national media. In addition
to HIPAA, most states have laws requiring notification of affected individuals and state regulators in the event of a breach of “personal
information,” which is a broader class of information than the PHI protected by HIPAA. Certain states also have data privacy requirements
applicable to individually identifiable health information. Privacy laws in different states may contain different requirements, and
such laws may not be pre-empted by HIPAA, which could complicate our efforts to comply.
In
addition, even when HIPAA does not apply, according to the FTC, failing to take appropriate steps to keep consumers’ personal information
secure constitutes unfair acts or practices in or affecting commerce in violation of Section 5(a) of the FTCA, 15 U.S.C § 45(a).
The FTC expects a company’s data security measures to be reasonable and appropriate in light of the sensitivity and volume of consumer
information it holds, the size and complexity of its business, and the cost of available tools to improve security and reduce vulnerabilities.
Medical data is considered sensitive data that merits stronger safeguards. The FTC’s guidance for appropriately securing consumers’
personal information is similar to what is required by the HIPAA Security Rule.
Our
failure to comply with applicable laws and regulations, or to protect such data, could result in enforcement actions against us, including
fines, imprisonment of company officials and public censure, claims for damages by end-customers, and other affected individuals, and
the imposition of integrity obligations and agency oversight, damage to our reputation, and loss of goodwill, any of which could harm
on our operations, financial performance, and business. Evolving and changing definitions of personal data and personal information,
within the United States, and elsewhere, may limit or inhibit our ability to operate or expand our business, including limiting strategic
partnerships that may involve the sharing of data. Moreover, if the relevant laws and regulations change, or are interpreted and applied
in a manner that is inconsistent with our data practices or the operation of our product, or if we expand into new regions and are required
to comply with new requirements, we may need to expend resources in order to change our business operations, data practices, or the manner
in which our product operates. Even the perception of privacy concerns, whether or not valid, may harm our reputation and inhibit adoption
of our product.
Even
if our product is approved by regulatory authorities if our contract manufacturers fail to comply with ongoing FDA, or if we experience
unanticipated problems with our products, these products could be subject to restrictions or withdrawal from the market.
Any
product for which we obtain regulatory clearance or approval, and the manufacturing processes, reporting requirements, post-approval
clinical data, and promotional activities for such product, will be subject to continued regulatory review, oversight and periodic inspections
by the FDA and other domestic bodies. In particular, we and our contract manufacturers are required to comply with FDA’s Quality
System Regulations (“QSR”) for the manufacture of our product and other regulations which cover the methods and documentation
of the design, testing, production, control, quality assurance, labeling, packaging, storage, and shipping of any product for which we
obtain regulatory clearance or approval.
The
failure by us or one of our contract manufacturers to comply with applicable statutes and regulations, or the failure to timely and adequately
respond to any adverse inspectional observations or product safety issues, could result in, among other things, any of the following
enforcement actions:
| ● | untitled
letters, warning letters, fines, injunctions, consent, and civil penalties; |
| ● | unanticipated
expenditures to address or defend such actions; |
| ● | customer
notifications for repair, replacement, refunds; |
| ● | recall,
detention, or seizure of our product; |
| ● | operating
restrictions or partial suspension or total shutdown of production; |
| ● | refusing
or delaying our requests for 510(k) clearance or premarket approval and conformity assessments of new products or modified products; |
| ● | limitations
on the intended uses for which the product may be marketed; |
| ● | withdrawing
510(k) clearances or PMA approvals that have already been granted; or |
In
addition, we may be required to conduct costly post-market testing and surveillance to monitor the safety or effectiveness of our product,
and we must comply with medical device reporting requirements, including the reporting of adverse events and malfunctions related to
our product. Later discovery of previously unknown problems with our product, including unanticipated adverse events or adverse events
of unanticipated severity or frequency, manufacturing problems, or failure to comply with regulatory requirements such as QSR, may result
in changes to labeling, restrictions on such products or manufacturing processes, withdrawal of the products from the market, voluntary
or mandatory recalls, a requirement to repair, replace, or refund the cost of any medical device we manufacture or distribute, fines,
suspension, variation, or withdrawal of regulatory approvals product seizures, injunctions, or the imposition of civil, administrative,
or criminal penalties which would adversely affect our business, operating results, and prospects.
If
the FDA determines that our promotional materials, labeling, training or other marketing or educational activities constitute promotion
of an unapproved use, it could request that we cease or modify our training or promotional materials or subject us to regulatory enforcement
actions. It is also possible that other federal, state or foreign enforcement authorities might take action if they consider our training
or other promotional materials to constitute promotion of an unapproved use, which could result in significant fines or penalties under
other statutory authorities, such as laws prohibiting false or fraudulent claims for payment of government funds.
If
any of these actions were to occur it would harm our reputation and cause our product sales and profitability to suffer and may prevent
us from generating revenue. Furthermore, our key component suppliers may not currently be or may not continue to be in compliance with
all applicable regulatory requirements, which could result in our failure to produce our product on a timely basis and in the required
quantities, if at all.
The
FDA has not yet inspected our facility, but we expect an inspection in the future.
Our
employees, independent contractors, consultants, contract manufacturers, and our independent sales representatives may engage in misconduct
or other improper activities, relating to regulatory standards and requirements.
We
are exposed to the risk that our employees, independent contractors, consultants, contract manufacturers, and our independent sales representatives
may engage in fraudulent conduct or other illegal activity. Misconduct by these parties could include intentional, reckless and/or negligent
conduct or disclosure of unauthorized activities to us that violates FDA regulations, including those laws requiring the reporting of
true, complete and accurate information to the FDA, manufacturing standards, federal and state healthcare laws and regulations, and laws
that require the true, complete and accurate reporting of financial information or data. These laws and regulations may restrict or prohibit
a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs, and other business arrangements.
Misconduct by these parties could also involve the improper use of individually identifiable information, including, without limitation,
information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation.
We plan to implement a compliance program, code of conduct and associated policies and procedures, but it is not always possible to identify
and deter misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or
unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to
be in compliance with such laws or regulations. If any such actions are instituted against us, and we are not successful in defending
ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant
civil, criminal, and administrative penalties, including, without limitation, damages, fines, disgorgement of profits, imprisonment,
exclusion from participation in government healthcare programs, such as Medicare and Medicaid, and the curtailment or restructuring of
our operations.
We
may be subject to enforcement action, including fines, penalties or injunctions, if we are determined to be engaging in the off-label
promotion of our product.
Our
promotional materials and training methods must comply with FDA and other applicable laws and regulations, including the prohibition
of the promotion of off-label use. Physicians may use our product off-label, as the FDA does not restrict or regulate a physician’s
choice of treatment within the practice of medicine. In the United States, the full indication for The Catamaran System is: “The
Tenon Catamaran Sacroiliac Joint Fixation System (CAT SIJ Fixation System) is intended for sacroiliac joint fusion for conditions including
sacroiliac joint disruptions and degenerative sacroiliitis.” Contraindications are patients with the following conditions: skeletally
immature spines; deformities; severe osteoporosis; morbid obesity, tumor resection and active infection at treatment site.
We
believe that the specific surgical procedures for which our product are marketed fall within the scope of the surgical applications that
have been cleared by the FDA. However, if the FDA determines that our promotional materials or training constitutes promotion of an off-label
use, it could request that we modify our training or promotional materials, require us to stop promoting our product for those specific
procedures until we obtain FDA clearance or approval for them, or subject us to regulatory or enforcement actions, including the issuance
of an untitled letter, a warning letter, injunction, seizure, civil fines, and criminal penalties. It is also possible that other federal,
state or foreign enforcement authorities might take action if they consider our promotional or training materials to constitute promotion
of an unapproved use, which could result in significant fines or penalties under other statutory authorities, such as laws prohibiting
false or fraudulent claims for payment of government fund. In that event, our reputation could be damaged, and adoption of the product
would be impaired. Although our policy is to refrain from statements that could be considered off-label promotion of our product, the
FDA or another regulatory agency could disagree and conclude that we have engaged in off-label promotion. In addition, the off-label
use of our product may increase the risk of injury to patients, and, in turn, the risk of product liability claims. Product liability
claims are expensive to defend and could divert our management’s attention, result in substantial damage awards against us and
harm our reputation.
We
are required to report certain malfunctions, deaths, and serious injuries associated with our product, which can result in voluntary
corrective actions or agency enforcement actions.
Further,
under the FDA’s medical device reporting regulations, we are required to report to the FDA any information that our product may
have caused or contributed to a death or serious injury or in which our product malfunctioned and, if the malfunction were to recur,
would likely cause or contribute to death or serious injury. If we fail to report these events to the FDA within the required timeframes,
or at all, FDA could take enforcement action against us. Any such adverse event involving our product or repeated product malfunctions
may result in a voluntary or involuntary corrective actions, such as recalls or customer notifications, or agency action, such as inspection
or enforcement action. Any corrective action, whether voluntary or involuntary, as well as defending ourselves in a lawsuit could divert
managerial and financial resources, impair our ability to manufacture our product in a cost-effective and timely manner, and have an
adverse effect on our reputation, results of operations, and financial condition.
Any
adverse event involving our product in the United States could result in future voluntary corrective actions, such as recalls, including
corrections, or customer notifications, or agency action, such as inspection or enforcement actions. If malfunctions do occur, we may
be unable to correct the malfunctions adequately or prevent further malfunctions, in which case we may need to cease manufacture and
distribution of the affected products, initiate voluntary recalls, and redesign the products. Regulatory authorities may also take actions
against us, such as ordering recalls, imposing fines, or seizing the affected products. Any corrective action, whether voluntary or involuntary,
will require the dedication of our time and capital, distract management from operating our business, and may harm our reputation and
financial results.
A
recall of our product, either voluntarily or at the direction of the FDA or the discovery of serious safety issues or malfunctions with
our product, can result in voluntary corrective actions or agency enforcement actions, which could have a significant adverse impact
on us.
The
FDA has the authority to require the recall of commercialized products in the event of material deficiencies or defects in design or
manufacture or in the event that a product poses an unacceptable risk to health. Manufacturers may, under their own initiative, recall
a product if any material deficiency in a device is found.
In
the case of the FDA, the authority to require a recall must be based on an FDA finding that there is an unreasonable risk of substantial
public harm. In addition, foreign governmental bodies have the authority to require the recall of our product in the event of material
deficiencies or defects in design or manufacture. A government-mandated or voluntary recall by us or one of the independent sales representatives
could occur as a result of an unacceptable risk to health, component failures, manufacturing errors, design or labeling defects, or other
deficiencies and issues. Recalls of any of our product would divert managerial and financial resources and have an adverse effect on
our reputation, results of operations, and financial condition, which could impair our ability to produce our product in a cost-effective
and timely manner in order to meet our customers’ demands. We may also be required to bear other costs or take other actions that
may have a negative impact on our future sales and our ability to generate profits.
The
FDA requires that certain classifications of recalls be reported to FDA within 10 working days after the recall is initiated. Companies
are required to maintain certain records of recalls, even if they are not reportable to the FDA. We may initiate voluntary recalls involving
our product in the future that we determine do not require notification of the FDA. If the FDA disagrees with our determinations, they
could require us to report those actions as recalls. A future recall announcement could harm our reputation with customers and negatively
affect our sales. In addition, the FDA could take enforcement action for failing to report the recalls when they were conducted.
Modifications
to our product may require new 510(k) clearances or premarket approvals may require us to cease marketing or recall the product until
clearances
Any
modification to a 510(k)-cleared device that could significantly affect its safety or effectiveness, or that would constitute a major
change in its intended use, design, or manufacture, requires a new 510(k) clearance or, possibly, a PMA. The FDA requires every manufacturer
make and document this determination in the first instance. A manufacturer may determine that a modification could not significantly
affect safety or effectiveness and does not represent a major change in its intended use, so that no new 510(k) clearance is necessary.
FDA may review any manufacturer’s decision and may not agree with our decisions regarding whether new clearances or approvals are
necessary. The FDA may also on its own initiative determine that a new clearance or approval is required.
We
have modified our product and have determined based on our review of the applicable FDA guidance that a new 510(k) clearances or PMAs
is not required. If the FDA disagrees with our determination and requires us to submit new 510(k) clearances or PMAs for modifications
to our previously cleared products for which we have concluded that new clearances or approvals are unnecessary, we may be required to
cease marketing or to recall the modified product until we obtain clearance or approval, and we may be subject to significant enforcement
action, regulatory fines, or penalties.
If
a manufacturer determines that a modification to an FDA-cleared device could significantly affect its safety or effectiveness or would
constitute a major change in its intended use, then the manufacturer must file for a new 510(k) clearance or possibly a premarket approval
application. Where we determine that modifications to our product require a new 510(k) clearance or premarket approval application, we
may not be able to obtain those additional clearances or approvals for the modifications or additional indications in a timely manner,
or at all. FDA’s ongoing review of the 510(k) programs may make it more difficult for us to make modifications to our previously
cleared products, either by imposing more strict requirements on when a new 510(k) for a modification to a previously cleared product
must be submitted or applying more onerous review criteria to such submissions.
Clinical
trials necessary to support a 510(k) or reimbursement may require the enrollment of large numbers of patients, and suitable patients
may be difficult to identify and recruit. Delays or failures in our clinical trials could affect third party reimbursement as many of
the payors want to see peer reviewed articles to maintain coverage and lack of changes in reimbursement could materially slow down our
commercial efforts and affect our revenue projections.
The
results of our clinical trials may not support our product candidate claims or may result in the discovery of adverse side effects.
If
our clinical trials are completed as planned, we cannot be certain that their results will support our product marketing claims or third
party reimbursors will agree with our conclusions regarding them. The clinical trial process may fail to demonstrate efficacy and cost
effectiveness of our product and may hinder the adoption of our product or ability to obtain payor coverage. It is also possible that
patients enrolled in clinical trials will experience adverse side effects that are not currently part of the product candidate’s
profile.
We
may incur product liability losses, and insurance coverage may be inadequate or unavailable to cover these losses.
Our
business exposes us to potential product liability claims that are inherent in the testing, design, manufacture, and sale of medical
devices for SI-Joint surgery procedures. SI-Joint surgery involves significant risk of serious complications, including bleeding, nerve
injury, paralysis, and even death. In addition, if longer-term patient results and experience indicates that our product or any component
of such product cause tissue damage, motor impairment, or other adverse effects, we could be subject to significant liability. Clinicians
may misuse or ineffectively use our product, which may result in unsatisfactory patient outcomes or patient injury. We could become the
subject of product liability lawsuits alleging that component failures, manufacturing flaws, design defects, or inadequate disclosure
of product-related risks or product-related information resulted in an unsafe condition or injury to patients. Product liability lawsuits
and claims, safety alerts, or product recalls, regardless of their ultimate outcome, could have a material adverse effect on our business
and reputation, our ability to attract and retain customers and our results of operations or financial condition.
Although
we maintain third-party product liability insurance coverage, it is possible that claims against us may exceed the coverage limits of
our insurance policies or cause us to record a self-insured loss. Even if any product liability loss is covered by an insurance policy,
these policies typically have substantial retentions or deductibles that we are responsible for. Product liability claims in excess of
applicable insurance coverage could have a material adverse effect on our business, results of operations, and financial condition.
In
addition, any product liability claim brought against us, with or without merit, could result in an increase of our product liability
insurance rates. Insurance coverage varies in cost and can be difficult to obtain, and we cannot guarantee that we will be able to obtain
insurance coverage in the future on terms acceptable to us or at all.
We
are subject to environmental laws and regulations that can impose significant costs and expose us to potential financial liabilities.
Our
business and facility and those of our contract manufacturer are subject to foreign, federal, state, and local laws and regulations relating
to the protection of human health and the environment, including those governing the use, manufacture, storage, handling, and disposal
of, and exposure to, such materials and wastes. In addition, under some environmental laws and regulations, we could be held responsible
for costs relating to any contamination at our past or present facilities and at third-party waste disposal sites even if such contamination
was not caused by us. A failure to comply with current or future environmental laws and regulations could result in severe fines or penalties.
Any such expenses or liability could have a significant negative impact on our business, results of operations, and financial condition.
U.S.
tax legislation may materially affect our financial condition, results of operations and cash flows.
The
Tax Cuts and Jobs Act (the “Tax Act”) has significantly changed the U.S. federal income taxation of U.S. businesses, including
by reducing the U.S. corporate income tax rate, limiting interest deductions, permitting immediate expensing of certain capital expenditures,
modifying or repealing many business deductions and credits.
The
Coronavirus Aid, Relief, and Economic Security Act (the “CARES Act”) modifies certain provisions of the Tax Act, including
increasing the amount of interest expense that may be deducted.
The
Tax Act as modified by the CARES Act is unclear in many respects and could be subject to potential amendments and technical corrections,
as well as interpretations and implementing regulations by the Treasury and IRS, any of which could lessen or increase certain adverse
impacts of the legislation. In addition, it is unclear how these U.S. federal income tax changes will affect state and local taxation,
which often uses federal taxable income as a starting point for computing state and local tax liabilities. Our analysis and interpretation
of this legislation is preliminary and ongoing and there may be material adverse effects resulting from the legislation that we have
not yet identified. While some of the changes made by the tax legislation may adversely affect us, other changes may be beneficial. We
continue to work with our tax advisors and auditors to determine the full impact that the recent tax legislation as a whole will have
on us. We urge our investors to consult with their legal and tax advisors with respect to such legislation and its potential effect on
an investment in our common stock.
Risks
Related to Our Intellectual Property
Our
ability to protect our intellectual property and proprietary technology is uncertain.
We
rely primarily on patent, copyright, trademark and trade secret laws, as well as confidentiality and non- disclosure agreements and other
methods, to protect our proprietary technologies and know-how. As of August 13, 2024, we owned eight issued patents (four domestic and
four foreign), 20 pending patent applications (18 domestic and two foreign), thirteen registered trademarks (seven domestic and six foreign)
and twelve pending domestic trademark applications.
We
have applied for patent protection relating to certain existing and proposed products and processes. While we generally apply for patents
in those countries where we intend to make, have made, use, or sell patented products, we may not accurately predict all the countries
where patent protection will ultimately be desirable. If we fail to timely file a patent application in any such country, we may be precluded
from doing so later. Furthermore, we cannot assure you that any of our patent applications will be approved. The rights granted to us
under our patents, including prospective rights sought in our pending patent applications, may not be meaningful or provide us with any
commercial advantage. In addition, those rights could be opposed, contested, or circumvented by our competitors or be declared invalid
or unenforceable in judicial or administrative proceedings. The failure of our patents to adequately protect our technology might make
it easier for our competitors to offer the same or similar products or technologies. Competitors may be able to design around our patents
or develop products that provide outcomes which are comparable to ours without infringing on our intellectual property rights. Due to
differences between foreign and U.S. patent laws, our patented intellectual property rights may not receive the same degree of protection
in foreign countries as they would in the United States. Even if patents are granted outside the United States, effective enforcement
in those countries may not be available. Since most of our issued patents are for the United States only, we lack a corresponding scope
of patent protection in other countries. In countries where we do not have significant patent protection, we may not be able to stop
a competitor from marketing products in such countries that are the same as or similar to our product.
We
plan to rely on our trademarks, trade names and brand names to distinguish our product from the products of our competitors and have
registered or applied to register many of these trademarks. We cannot assure you that our trademark applications will be approved. Third
parties may also oppose our trademark applications, or otherwise challenge our use of the trademarks. In the event that our trademarks
are successfully challenged, we could be forced to rebrand our product, which could result in loss of brand recognition, and could require
us to devote resources to advertising and marketing new brands. Further, we cannot assure you that competitors will not infringe upon
our trademarks, or that we will have adequate resources to enforce our trademarks.
We
also rely on trade secrets, know-how, and technology, which are not protected by patents, to maintain our competitive position. We try
to protect this information by entering into confidentiality and intellectual property assignment agreements with parties that develop
intellectual property for us and/or have access to it, such as our officers, employees, consultants, contract manufacturers and advisors.
However, in the event of unauthorized use or disclosure or other breaches of such agreements, we may not be provided with meaningful
protection for our trade secrets or other proprietary information. In addition, our trade secrets may otherwise become known or be independently
discovered by competitors. To the extent that our commercial partners, collaborators, employees, and consultants use intellectual property
owned by others in their work for us, disputes may arise as to the rights in related or resulting know-how and inventions. If any of
our trade secrets, know-how or other technologies not protected by a patent were to be disclosed to or independently developed by a competitor,
our business, financial condition, and results of operations could be materially adversely affected.
In
the future, we may enter into licensing agreements to maintain our competitive position. If we enter into in-bound intellectual property
license agreements, we may not be able to fully protect the licensed intellectual property rights or maintain those licenses. Future
licensors could retain the right to prosecute and defend the intellectual property rights licensed to us, in which case we would depend
on the ability of our licensors to obtain, maintain, and enforce intellectual property protection for the licensed intellectual property.
These licensors may determine not to pursue litigation against other companies or may pursue such litigation less aggressively than we
would. Further, entering into such license agreements could impose various diligence, commercialization, royalty, or other obligations
on us. Future licensors may allege that we have breached our license agreement with them, and accordingly seek damages or to terminate
our license, which could adversely affect our competitive business position and harm our business prospects.
If
a competitor infringes upon one of our patents, trademarks, or other intellectual property rights, enforcing those patents, trademarks,
and other rights may be difficult and time consuming. Even if successful, litigation to defend our patents and trademarks against challenges
or to enforce our intellectual property rights could be expensive and time consuming and could divert management’s attention from
managing our business. Moreover, we may not have sufficient resources to defend our patents or trademarks against challenges or to enforce
our intellectual property rights. In addition, if third parties infringe any intellectual property that is not material to the products
that we make, have made, use, or sell, it may be impractical for us to enforce this intellectual property against those third parties.
We
may be subject to damages resulting from claims that we, our employees, or independent distributors along with their independent sales
representatives have wrongfully used or disclosed alleged trade secrets of our competitors or are in breach of non-competition or non-solicitation
agreements with our competitors.
Many
of our employees were previously employed at other medical device companies, including our competitors or potential competitors, in some
cases until recently. Some independent distributors and their independent sales representatives sell, or in the past have sold, products
of our competitors. We may be subject to claims that we, our employees or independent sales personnel have inadvertently or otherwise
used or disclosed trade secrets or other proprietary information of these former employers or competitors. In addition, we have been
and may in the future be subject to claims that we caused an employee to breach the terms of his or her non-competition or non-solicitation
agreement. Even if we are successful in defending against these claims, litigation could result in substantial costs, divert the attention
of management from our core business and harm our reputation. If our defense to those claims fails, in addition to paying monetary damages,
we may lose valuable intellectual property rights or personnel. There can be no assurance that this type of litigation will not continue,
and any future litigation or the threat thereof may adversely affect our ability to hire additional direct sales representatives. A loss
of key personnel or their work product could hamper or prevent our ability to commercialize product candidates, which could have an adverse
effect on our business, results of operations, and financial condition.
The
medical device industry is characterized by patent litigation, and we could become subject to litigation that could be costly, result
in the diversion of management’s time and efforts, require us to pay damages, and/or prevent us from developing or marketing our
existing or future products.
Our
commercial success will depend in part on not infringing the patents or violating the other proprietary rights of third parties. Significant
litigation regarding patent rights exists in our industry. Our competitors in both the United States and abroad, many of which have substantially
greater resources and have made substantial investments in competing technologies, may have applied for or obtained or may in the future
apply for and obtain, patents that will prevent, limit, or otherwise interfere with our ability to make and sell our product. We have
conducted a limited review of patents issued to third parties. The large number of patents, the rapid rate of new patent issuances, the
complexities of the technology involved, and the uncertainty of litigation increase the risk of business assets and management’s
attention being diverted to patent litigation. Any litigation or claim against us, even those without merit, may cause us to incur substantial
costs, and could place a significant strain on our financial resources, divert the attention of management from our core business, and
harm our reputation. Further, as the number of participants in the medical device industry grows, the possibility of intellectual property
infringement claims against us increases. If we are found to infringe the intellectual property rights of third parties, we could be
required to pay substantial damages, including treble, or triple, damages if an infringement is found to be willful, and/or royalties
and could be prevented from selling our product unless we obtain a license or are able to redesign our product to avoid infringement.
Any such license may not be available on reasonable terms, if at all, and there can be no assurance that we would be able to redesign
our product in a way that would not infringe the intellectual property rights of others. If we fail to obtain any required licenses or
make any necessary changes to our product or technologies, we may have to withdraw our existing product from the market or may be unable
to commercialize one or more of our future products, all of which could have a material adverse effect on our business, results of operations,
and financial condition. If passed into law, patent reform legislation currently pending in the U.S. Congress could significantly change
the risks associated with bringing or defending a patent infringement lawsuit. For example, fee shifting legislation could require a
non-prevailing party to pay the attorney fees of the prevailing party in some circumstances.
Patent
terms are limited, and we may not be able to effectively protect our product and business.
Patents
have a limited lifespan. In the U.S., the natural expiration of a patent is generally 20 years after it is filed. Although various extensions
may be available, the life of a patent, and the protection it affords, is limited. In addition, upon issuance in the U.S., the patent
term may be extended based on certain delays caused by the applicant(s) or the USPTO. Even if we obtain effective patent rights for all
our current patent applications, we may not have sufficient patent terms or regulatory exclusivity to protect our product, and our business
and results of operations would be adversely affected.
Changes
in U.S. patent law could diminish the value of patents in general, thereby impairing our ability to protect our product.
As
is the case with other medical devices companies, our success is heavily dependent on intellectual property, particularly patents. Obtaining
and enforcing patents in the medical devices industry involves both technological and legal complexity. Therefore, obtaining and enforcing
patents is costly, time-consuming, and inherently uncertain. In addition, the U.S. has recently enacted and is currently implementing
wide-ranging patent reform legislation. Recent U.S. Supreme Court rulings have narrowed the scope of patent protection available in certain
circumstances and weakened the rights of patent owners in certain situations. In addition to increasing uncertainty with regard to our
ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents, once
obtained. Depending on future actions by the U.S. Congress, the federal courts and the USPTO, the laws and regulations governing patents
could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce our existing patents and patents
that we might obtain in the future.
We
may not be able to protect our intellectual property rights throughout the world.
Filing,
prosecuting, and defending patents on product candidates in all countries throughout the world would be prohibitively expensive, and
our intellectual property rights in some countries outside the U.S. can be less extensive than those in the U.S. In addition, the laws
of some foreign countries do not protect intellectual property rights to the same extent as federal and state laws in the U.S. Competitors
may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and may also export
otherwise infringing products to territories where we have patent protection, but enforcement is not as strong as that in the U.S. These
products may compete with our product and our patents or other intellectual property rights may not be effective or sufficient to prevent
them from competing.
Many
companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The
legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents, trade secrets
and other intellectual property protection, particularly those relating to biotechnology products, which could make it difficult for
us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally. Proceedings
to enforce our patent rights in foreign jurisdictions, whether or not successful, could result in substantial costs and divert our efforts
and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly and our
patent applications at risk of not issuing and could provoke third parties to assert claims against us. We may not prevail in any lawsuits
that we initiate, and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to
enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual
property that we develop or license.
If
we are unable to protect the confidentiality of our trade secrets, our business and competitive position could be harmed.
In
addition to patent protection, we also rely upon copyright and trade secret protection, as well as non-disclosure agreements and invention
assignment agreements with our employees, consultants, contract manufacturers and third parties, to protect our confidential and proprietary
information. In addition to contractual measures, we try to protect the confidential nature of our proprietary information using commonly
accepted physical and technological security measures. Such measures may not, for example, in the case of misappropriation of a trade
secret by an employee or third party with authorized access, provide adequate protection for our proprietary information. Our security
measures may not prevent an employee or consultant from misappropriating our trade secrets and providing them to a competitor, and recourse
we take against such misconduct may not provide an adequate remedy to protect our interests fully. Unauthorized parties may also attempt
to copy or reverse engineer certain aspects of our product that we consider proprietary. Enforcing a claim that a party illegally disclosed
or misappropriated a trade secret can be difficult, expensive, and time-consuming, and the outcome is unpredictable. Even though we use
commonly accepted security measures, trade secret violations are often a matter of state law, and the criteria for protection of trade
secrets can vary among different jurisdictions. In addition, trade secrets may be independently developed by others in a manner that
could prevent legal recourse by us. If any of our confidential or proprietary information, such as our trade secrets, were to be disclosed
or misappropriated, or if any such information was independently developed by a competitor, our business and competitive position could
be harmed.
Third
parties may assert that our employees or consultants have wrongfully used or disclosed confidential information or misappropriated trade
secrets.
We
employ individuals who previously worked with other companies, including our competitors or potential competitors. Although we try to
ensure that our employees and consultants do not use the proprietary information or know-how of others in their work for us, we may be
subject to claims that we or our employees, consultants or independent contractors have inadvertently or otherwise used or disclosed
intellectual property, including trade secrets or other proprietary information, of a former employer or other third party. Litigation
may be necessary to defend against these claims. If we fail in defending any such claims or settling those claims, in addition to paying
monetary damages or a settlement payment, we may lose valuable intellectual property rights or personnel. Even if we are successful in
defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.
Risks
Related to the Offering and the Ownership of our Common Stock
This
is a reasonable best efforts offering, with no minimum amount of securities required to be sold, and we may sell fewer than all of the
securities offered hereby.
The
placement agent has agreed to use its reasonable best efforts to solicit offers to purchase the securities in this offering. The placement
agent has no obligation to buy any of the securities from us or to arrange for the purchase or sale of any specific number or dollar
amount of the securities. There is no required minimum number of securities that must be sold as a condition to completion of this offering.
The success of this offering will impact our ability to use the proceeds to execute our business plans. We may have insufficient capital
to implement our business plans and satisfy current obligations, potentially resulting in greater operating losses or dilution unless
we are able to raise the required capital from alternative sources. There is no assurance that alternative capital, if needed, would
be available on terms acceptable to us, or at all.
You
will experience immediate dilution in the net tangible book value per share of the common stock you purchase, and may experience additional
dilution in the future.
The
effective public offering price per share will be substantially higher than the pro forma net tangible book value per share of the outstanding
shares of our common stock immediately after this offering. Based on a assumed combined public offering price of $[*] per share and accompanying
Common Warrant, and our net tangible book value as of June 30, 2024, if you purchase our securities (assuming no sale of Pre-funded Warrants)
in this offering you will pay more for your shares of common stock than the amounts paid by our existing shareholders for their shares
of common stock and you will suffer immediate dilution of approximately $[*] per share in pro forma net tangible book value. In addition,
the shares of common stock issuable upon the exercise of the Common Warrants to be issued pursuant to the offering will further dilute
the ownership interest of shareholders not participating in this offering and holders of Warrants who have not exercised their Warrants.
As a result of this dilution, investors purchasing securities in this offering may receive significantly less than the full purchase
price that they paid for the shares of common stock purchased in this offering in the event of a liquidation. To the extent shares of
common stock are issued under outstanding convertible preferred stock, options and warrants at exercise prices lower than the public
offering price of our shares of common stock in this offering, including the shares of common stock underlying the Pre-Funded Warrants,
holders will incur further dilution.
We
have broad discretion in the use of the net proceeds we receive from this offering and may not use them effectively.
Our
management will have broad discretion in the application of the net proceeds we receive in this offering, including for any of the purposes
described in the section entitled “Use of Proceeds,” and you will not have the opportunity as part of your investment decision
to assess whether our management is using the net proceeds appropriately. Because of the number and variability of factors that will
determine our use of our net proceeds from this offering, their ultimate use may vary substantially from their currently intended use.
The failure by our management to apply these funds effectively could result in financial losses that could have a material adverse effect
on our business and cause the price of our common stock to decline. Pending their use, we may invest our net proceeds from this offering
in short-term, investment-grade, interest-bearing securities. These investments may not yield a favorable return to our stockholders.
An
active trading market for our shares may not be sustained.
Although
our shares are listed on The Nasdaq Stock Market LLC, the market for our shares has demonstrated varying levels of trading activity.
The current level of trading may not be sustained in the future. The lack of an active market for our shares may impair investors’
ability to sell their shares at the time they wish to sell them or at a price that they consider reasonable, may reduce the fair market
value of their shares and may impair our ability to raise capital to continue to fund operations by selling shares and may impair our
ability to acquire additional intellectual property assets by using our shares as consideration.
This
offering may cause the trading price of our common stock to decrease.
The
number of shares of common stock underlying the securities we propose to issue and ultimately will issue if this offering is completed,
may result in an immediate decrease in the market price of our common stock. This decrease may continue after the completion of this
offering. We cannot predict the effect, if any, that the availability of shares for future sale represented by the Pre-funded Warrants
or Common Warrants issued in connection with the offering will have on the market price of our common stock from time to time.
Future
sales of substantial amounts of our common stock could adversely affect the market price of our common stock.
We
may choose to raise additional capital due to market conditions or strategic considerations even if we believe we have sufficient funds
for our current or future operating plans. If additional capital is raised through the sale of equity or convertible debt securities,
or perceptions that those sales could occur, the issuance of these securities could result in further dilution to investors purchasing
our common stock in this offering or result in downward pressure on the price of our common stock, and our ability to raise capital in
the future. We currently have a facility with Lincoln Park Capital Fund, LLC (“Lincoln Park”) in which we may sell up to
$10 million in share of common stock to Lincoln Park. As of the date of the prospectus, we have issued and sold [*] shares to Lincoln
Park all of which have been registered, we have effective registration statements that cover sales of [*] additional shares to Lincoln
Park.
We
have previously received a notice from Nasdaq that we were not in compliance with the Nasdaq continued listing requirements. If we are
unable to regain compliance with Nasdaq’s listing requirements, our common stock could be delisted, which could affect our common
stock’s market price and liquidity and reduce our ability to raise capital.
On
May 7, 2024, we received a letter from the Nasdaq Listing Qualifications Staff of Nasdaq stating that for the 30 consecutive business
day period between March 25, 2024 and May 6, 2024, our common stock had not maintained a minimum closing bid price of $1.00 per share
required for continued listing on The Nasdaq Capital Market pursuant to Nasdaq Listing Rule 5550(a)(2) (the “Bid Price Rule”).
Pursuant to Nasdaq Listing Rule 5810(c)(3)(A), we were provided an initial period of 180 calendar days, or until November 4, 2024 (the
“Compliance Period”), to regain compliance with the Bid Price Rule.
To
regain compliance, the closing bid price of our common stock must meet or exceed $1.00 per share for a minimum of 10 consecutive trading
days, unless extended by Nasdaq under Nasdaq Rule 5810(c)(3)(H), prior to November 4, 2024.
If
we are unable to maintain compliance with such listing standards or other Nasdaq listing requirements in the future, we could be subject
to suspension and delisting proceedings. A delisting of our common stock and our inability to list on another national securities market
could negatively impact us by: (i) reducing the liquidity and market price of our common stock; (ii) reducing the number of investors
willing to hold or acquire our common stock, which could negatively impact our ability to raise equity financing; (iii) limiting our
ability to use certain registration statements to offer and sell freely tradable securities, thereby limiting our ability to access the
public capital markets; and (iv) impairing our ability to provide equity incentives to our employees.
There
is no public market for the Common Warrants or Pre-Funded Warrants being offered by us in this offering.
There
is no established public trading market for the Common Warrants or the Pre-Funded Warrants, and we do not expect a market to develop.
In addition, we do not intend to apply to list the Warrants or the Pre-Funded Warrants on any national securities exchange or other nationally
recognized trading system. Without an active market, the liquidity of the Warrants and the Pre-Funded Warrants will be limited.
The
Common Warrants are speculative in nature.
The
Common Warrants will be exercisable for five years from the date of initial exercise at an exercise price of $[*] per share (assuming
an exercise price equal to 100% of the assumed public offering price per share). There can be no assurance that the market price of the
common stock will ever equal or exceed the exercise price of the Common Warrants. In the event that our common stock price does not exceed
the exercise price of the Common Warrants during the period when the Common Warrants are exercisable, a holder of the Common Warrants
may be unable to profit from exercising such Common Warrants before they expire.
Except
as otherwise provided in the Common Warrants and Pre-funded Warrants, holders of Common Warrants and Pre-funded Warrants purchased in
this offering will have no rights as stockholders until such holders exercise their Common Warrants or Pre-funded Warrants and acquire
our common stock.
Except
as otherwise provided in the Common Warrants and Pre-funded Warrants, until holders of Common Warrants or Pre-funded Warrants acquire
our common stock upon exercise of the Common Warrants or Pre-funded Warrants, holders of Common Warrants and Pre-funded warrants will
have no rights with respect to our common stock underlying such Common Warrants and Pre-funded Warrants. Upon exercise of the Common
Warrants and Pre-funded Warrants, the holders will be entitled to exercise the rights of a holder of our common stock only as to matters
for which the record date occurs after the exercise date.
Since
we do not expect to pay any cash dividends for the foreseeable future, investors may be forced to sell their stock in order to obtain
a return on their investment.
We
do not anticipate declaring or paying in the foreseeable future any cash dividends on our capital stock. Instead, we plan to retain any
earnings to finance our operations and growth plans discussed elsewhere or incorporated by reference in this prospectus. Accordingly,
investors must rely on sales of their common stock after price appreciation, which may never occur, as the only way to realize any return
on their investment. As a result, investors seeking cash dividends should not purchase our common stock.
The
trading price of our common stock has been and is likely to continue to be highly volatile and could be subject to wide fluctuations
in response to various factors, some of which are beyond our control.
Our
share price is highly volatile. During the period from August 1, 2023, to [*], 2024 the closing price of our common stock ranged from
a high of $[*] per share to a low of $[*] per share. The stock market in general has experienced extreme volatility that has often been
unrelated to the operating performance of particular companies. As a result of this volatility, you may not be able to sell your common
stock at or above the public offering price and you may lose some or all of your investment.
Our
Series A Preferred Stock ranks senior to our common stock.
Our
Series A Preferred Stock ranks, with respect to dividend rights, rights on the distribution of assets on any voluntary or involuntary
liquidation, dissolution or winding up of the affairs of our company, and redemption rights, senior to our common stock and each other
class or series of securities now existing or hereafter authorized classified or reclassified, the terms of which do not expressly provide
that such class or series ranks on a parity basis with or senior to the Series B Preferred Stock as to dividend rights, rights on the
distribution of assets on any voluntary or involuntary liquidation, dissolution or winding up of the affairs of our company, and redemption
rights.
Conversion
of Series A Preferred Stock or the exercise of the Tradeable Warrants or the Series A Warrants may cause significant dilution to our
stockholders.
As
of August 13, 2024 we have issued and outstanding 256,968 shares of Series A Preferred Stock, which are convertible into 2,569,680 shares
of common stock; Tradeable Warrants with 2,000,000 underlying shares of common stock; Note Warrants with 45,000 underlying shares of
common stock and Series A Warrants with 415,468 underlying shares of common stock. The issuance of shares of common stock upon the conversion
of such shares of preferred stock or exercise of any of such warrants would dilute the percentage ownership interest of holders of our
common stock, dilute the book value per share of our common stock and increase the number of our publicly traded shares, which could
depress the market price of our Common Stock.
In
addition, the Series A Warrants and the Note Warrants contain weighted average anti-dilution provisions which, subject to limited exceptions,
would increase the number of shares issuable upon conversion of such securities (by reducing the exercise price) in the event that we
in the future issue common stock, or securities convertible into or exercisable to purchase common stock, at a price per share lower
than the conversion price then in effect.
If
securities or industry analysts do not publish research or publish inaccurate or unfavorable research about our business, our stock price
and trading volume could decline.
The
trading market for our common stock will depend in part on the research and reports that securities or industry analysts publish about
us or our business. If too few securities or industry analysts provide coverage or if one or more of the analysts who cover us downgrade
our stock or publish inaccurate or unfavorable research about our business, the price of our stock would likely decline. If one or more
of these analysts cease coverage of us or fail to publish reports on us regularly, demand for our stock could decrease, which might cause
the price of our stock and trading volume to decline.
The
price of our common stock may be volatile, and you may be unable to resell your shares at or above the price paid.
The
trading price of our common stock may fluctuate substantially. The market price of our common stock may fluctuate higher or lower, depending
on many factors, some of which are beyond our control and may not be related to our operating performance. These fluctuations could cause
you to lose all or part of your investment in our common stock. Factors that could cause fluctuations in the trading price of our common
stock include the following:
| ● | actual
or anticipated fluctuations in our financial condition and operating results; |
| ● | actual
or anticipated changes in our growth rate relative to our competitors; |
| ● | commercial
success and market acceptance of our product; |
| ● | success
of our competitors in developing or commercializing products; |
| ● | ability
to commercialize or obtain regulatory approvals for our product, or delays in commercializing
or obtaining regulatory approvals; |
| ● | strategic
transactions undertaken by us; |
| ● | additions
or departures of key personnel; |
| ● | product
liability claims; |
| ● | prevailing
economic conditions; |
| ● | disputes
concerning our intellectual property or other proprietary rights; |
| ● | FDA
or other U.S. or foreign regulatory actions affecting us or the healthcare industry; |
| ● | healthcare
reform measures in the United States; |
| ● | sales
of our common stock by our officers, directors or significant stockholders; |
| ● | future
sales or issuances of equity or debt securities by us; |
| ● | business
disruptions caused by earthquakes, fires or other natural disasters; |
| ● | the
exercise and sale of any outstanding warrants or options; |
| ● | issuance
of new or changed securities analysts’ reports or recommendations regarding us; |
| ● | changes
in our capital structure, such as future issuances of debt or equity securities; |
| ● | short
sales, hedging and other derivative transactions involving our capital stock; and |
| ● | general
economic and geopolitical conditions, including the current or anticipated impact of military
conflict and related sanctions imposed on Russia by the United States and other countries
due to Russia’s invasion of Ukraine. |
In
addition, if the market for medical device or healthcare stocks or the stock market, in general, experience a loss of investor confidence,
the trading price of our common stock could decline for reasons unrelated to our business, results of operations, or financial condition.
The trading price of our common stock might also decline in reaction to events that affect other companies in our industry even if these
events do not directly affect us. In the past, following periods of volatility in the market price of a company’s securities, securities
class action litigation has often been brought against that company. If our stock price is volatile, we may become the target of securities
litigation. Securities litigation could result in substantial costs and divert our management’s attention and resources from our
business. This could have a material adverse effect on our business, results of operations, and financial condition.
Our
failure to maintain effective internal controls over financial reporting could have an adverse impact on us.
We
are required to establish and maintain appropriate internal controls over financial reporting. Failure to establish those controls, or
any failure of those controls once established, could adversely impact our public disclosures regarding our business, financial condition,
or results of operations. In addition, management’s assessment of internal controls over financial reporting may identify weaknesses
and conditions that need to be addressed in our internal controls over financial reporting or other matters that may raise concerns for
investors. Any actual or perceived weaknesses and conditions that need to be addressed in our internal control over financial reporting,
disclosure of management’s assessment of our internal controls over financial reporting or disclosure of our public accounting
firm’s attestation to or report on management’s assessment of our internal controls over financial reporting may have an
adverse impact on the price of our common stock.
A
control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of
the control system are met. In addition, the design of a control system must reflect the fact that there are resource constraints, and
the benefit of controls must be relative to their costs. Because of the inherent limitations in all control systems, no system of controls
can provide absolute assurance that all control issues and instances of fraud, if any, within our Company have been detected. These inherent
limitations include the realities that judgments in decision-making can be faulty and that breakdowns can occur because of simple error
or mistake. Further, controls can be circumvented by individual acts of some persons, by collusion of two or more persons, or by management
override of the controls. The design of any system of controls is also based in part upon certain assumptions about the likelihood of
future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions.
Over time, a control may become inadequate because of changes in conditions or the degree of compliance with policies or procedures may
deteriorate. Because of inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and may
not be detected.
At present, management has identified a material
weakness due to lack of segregation of duties. The lack of segregation of duties existed as a result of the Company having no employees
until June 2021. Management has taken steps to remedy this weakness by its hiring of a Chief Financial Officer, a director of SEC reporting
and compliance, a senior accountant, a cost accountant and external financial consultants, and plans to continue to add additional resources,
technology and headcount as warranted by the growth of the Company. We are in the process of putting proper policies and procedures in
place to ensure proper documentation is established and maintained for transactions that the Company enters into. On May 20, 2024, it
was announced that our Chief Financial Officer was retiring effective July 31, 2024 while continuing to assist the Company as Chief Financial
Officer Advisor. We are actively searching for a qualified candidate to fill the role of Chief Financial Officer. The Senior Director
of SEC reporting and compliance is our interim principal financial officer as of the date of this prospectus. While we believe these efforts
will improve our internal controls and address the underlying causes of the material weakness, such material weakness will not be remediated
until our remediation plan has been fully implemented and we have concluded that our controls are operating effectively for a sufficient
period of time. We cannot be certain that the steps we are taking will be sufficient to remediate the control deficiencies that led to
our material weakness in our internal control over financial reporting or prevent future material weaknesses or control deficiencies from
occurring. While we are working to remediate the material weakness as timely and efficiently as possible, at this time we cannot provide
an estimate of costs expected to be incurred in connection with the implementation of this remediation plan, nor can we provide an estimate
of the time it will take to complete this remediation plan. Even if management does establish effective remedial measures, we cannot guarantee
that those internal controls and disclosure controls that we put in place will prevent all possible errors, mistakes, or all fraud.
Our
financial controls and procedures may not be sufficient to ensure timely and reliable reporting of financial information, which, as a
public company, could materially harm our stock price.
We
will require significant financial resources to maintain our public reporting status. We cannot assure you we will be able to maintain
adequate resources to ensure that we will not have any future material weakness in our system of internal controls. The effectiveness
of our controls and procedures may in the future be limited by a variety of factors including:
| ● | faulty
human judgment and simple errors, omissions or mistakes; |
| ● | fraudulent
action of an individual or collusion of two or more people; |
| ● | inappropriate
management override of procedures; and |
| ● | the
possibility that any enhancements to controls and procedures may still not be adequate to
assure timely and accurate financial information. |
Our
internal control over financial reporting will be a process designed to provide reasonable assurance regarding the reliability of financial
reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles
in the United States of America. Our internal control over financial reporting includes those policies and procedures that (i) pertain
to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets
of the Company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements
in accordance with generally accepted accounting principles, and that receipts and expenditures of the Company are being made only in
accordance with authorizations of management and directors of the Company; and (iii) provide reasonable assurance regarding prevention
or timely detection of unauthorized acquisition, use, or disposition of the Company’s assets that could have a material effect
on the financial statements.
Despite
these anticipated controls, because of its inherent limitations, internal control over financial reporting may not prevent or detect
misstatements. Therefore, even those systems determined to be effective can provide only reasonable assurance of achieving their control
objectives. Furthermore, smaller reporting companies like us face additional limitations. Smaller reporting companies employ fewer individuals
and can find it difficult to employ resources for complicated transactions and effective risk management. Additionally, smaller reporting
companies tend to utilize general accounting software packages that lack a rigorous set of software controls.
If
we fail to have effective controls and procedures for financial reporting in place, we could be unable to provide timely and accurate
financial information and be subject to investigation by the SEC and civil or criminal sanctions.
We
must implement additional and expensive procedures and controls in order to grow our business and organization and to satisfy reporting
requirements, which will increase our costs and require additional management resources.
As
a public company, we are required to comply with the Sarbanes-Oxley Act of 2002 (the “Sarbanes-Oxley Act”) and the related
rules and regulations of the SEC, including the requirements that we maintain disclosure controls and procedures and adequate internal
control over financial reporting. Compliance with the Sarbanes-Oxley Act and other SEC and national exchange requirements will increase
our costs and require additional management resources. We have begun the process of upgrading our procedures and controls and will need
to begin implementing additional procedures and controls as we grow our business and organization and to satisfy new reporting requirements.
If we are unable to complete the required assessment as to the adequacy of our internal control over financial reporting, as required
by Section 404 of the Sarbanes-Oxley Act or if we fail to establish and maintain internal control over financial reporting, our ability
to produce timely, accurate and reliable periodic financial statements could be impaired.
If
we do not establish and maintain adequate internal control over financial reporting, investors could lose confidence in the accuracy
of our periodic reports filed under the Exchange Act. Additionally, our ability to obtain additional financing could be impaired or a
lack of investor confidence in the reliability and accuracy of our public reporting could cause our stock price to decline.
We
may be subject to securities litigation, which is expensive and could divert our management’s attention.
The
market price of our securities may be volatile, and in the past companies that have experienced volatility in the market price of their
securities have been subject to securities class action litigation. We may be the target of this type of litigation in the future. Securities
litigation against us could result in substantial costs and divert our management’s attention from other business concerns.
We
are an “emerging growth company” under the JOBS Act of 2012 and we cannot be certain if the reduced disclosure requirements
applicable to emerging growth companies will make our common stock less attractive to investors.
We
are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012 (the “JOBS Act”),
and we may take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that
are not “emerging growth companies” including, but not limited to, not being required to comply with the auditor attestation
requirements of Section 404 of the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation in our periodic
reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and
shareholder approval of any golden parachute payments not previously approved. We cannot predict if investors will find our common stock
less attractive because we may rely on these exemptions. If some investors find our common stock less attractive as a result, there may
be a less active trading market for our common stock and our stock price may be more volatile.
In
addition, Section 107 of the JOBS Act also provides that an “emerging growth company” can take advantage of the extended
transition period provided in Section 7(a)(2)(B) of the Securities Act of 1933 (the “Securities Act”) for complying with
new or revised accounting standards. In other words, an “emerging growth company” can delay the adoption of certain accounting
standards until those standards would otherwise apply to private companies. We are choosing to take advantage of the extended transition
period for complying with new or revised accounting standards.
We
will remain an “emerging growth company” until the last day of the fiscal year following the fifth anniversary of the date
of the first sale of our common stock pursuant to an effective registration statement under the Securities Act, although we will lose
that status sooner if our revenues exceed $1.235 billion, if we issue more than $1 billion in non-convertible debt in a three year period,
or we are deemed to be a large accelerated filer under applicable SEC rules.
Our
status as an “emerging growth company” under the JOBS Act may make it more difficult to raise capital as and when we need
it.
Because
of the exemptions from various reporting requirements provided to us as an “emerging growth company” and because we will
have an extended transition period for complying with new or revised financial accounting standards, we may be less attractive to investors,
and it may be difficult for us to raise additional capital as and when we need it. Investors may be unable to compare our business with
other companies in our industry if they believe that our financial accounting is not as transparent as other companies in our industry.
If we are unable to raise additional capital as and when we need it, our financial condition and results of operations may be materially
and adversely affected.
We
have not paid dividends in the past and do not expect to pay dividends in the future, and any return on investment may be limited to
the value of our stock.
We
have never paid cash dividends on our common stock and do not anticipate paying cash dividends on our common stock in the foreseeable
future. We currently intend to retain any future earnings to support the development of our business and do not anticipate paying cash
dividends in the foreseeable future. Our payment of any future dividends will be at the discretion of our Board after taking into account
various factors, including, but not limited to, our financial condition, operating results, cash needs, growth plans and the terms of
any credit agreements that we may be a party to at the time. In addition, our ability to pay dividends on our common stock may be limited
by Delaware state law. Accordingly, investors must rely on sales of their common stock after price appreciation, which may never occur,
as the only way to realize a return on their investment. Investors seeking cash dividends should not purchase our common stock.
The
elimination of personal liability against our directors and officers under Delaware law and the existence of indemnification rights held
by our directors, officers and employees may result in substantial expenses.
Our
amended and restated certificate of incorporation, as amended (“Certificate of Incorporation”), and our bylaws (“Bylaws”)
eliminate the personal liability of our directors and officers to us and our stockholders for damages for breach of fiduciary duty as
a director or officer to the extent permissible under Delaware law. Further, our Certificate of Incorporation allows for us to and our
Bylaws provide that we are obligated to indemnify each of our directors or officers to the fullest extent authorized by Delaware law
and, subject to certain conditions, advance the expenses incurred by any director or officer in defending any action, suit or proceeding
prior to its final disposition. Those indemnification obligations could expose us to substantial expenditures to cover the cost of settlement
or damage awards against our directors or officers, which we may be unable to afford. Further, those provisions and resulting costs may
discourage us or our stockholders from bringing a lawsuit against any of our current or former directors or officers for breaches of
their fiduciary duties, even if such actions might otherwise benefit our stockholders.
Our
Certificate of Incorporation will designate the Court of Chancery of the State of Delaware as the exclusive forum for certain litigation
that may be initiated by our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for
disputes with us.
Our
Certificate of Incorporation specifies that, unless we consent in writing to the selection of an alternative forum, the Court of Chancery
of the State of Delaware shall be the sole and exclusive forum for (a) any derivative action or proceeding brought on behalf of the Company,
(b) any action asserting a claim of breach of a fiduciary duty owed by any director, officer, employee or agent of the Company to the
Company or the Company’s stockholders, (c) any action asserting a claim arising pursuant to any provision of the Delaware General
Corporation Law, our Certificate of Incorporation or Bylaws, or (d) any action asserting a claim governed by the internal affairs doctrine,
in each case subject to said Court of Chancery having personal jurisdiction over the indispensable parties named as defendants therein.
However, prior to the effectiveness of the registration statement related to this prospectus, we will amend our Certificate of Incorporation
to include a statement that this exclusive forum provision does not apply to claims arising under federal securities laws. Any person
or entity purchasing or otherwise acquiring any interest in shares of our capital stock shall be deemed to have notice of and to have
consented to the provisions of our Certificate of Incorporation as described above.
This
choice of forum provision may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes
with us or any of our directors, officers, other employees or stockholders, which may discourage lawsuits with respect to such claims.
As such, stockholders of the Company seeking to bring a claim regarding the internal affairs of the Company may be subject to increased
costs associated with litigating in Delaware as opposed to their home state or other forum, precluded from bringing such a claim in a
forum they otherwise consider to be more favorable, and discouraged from bringing such claims as a result of the foregoing or other factors
related to forum selection. Alternatively, if a court were to find the choice of forum provision contained in our Certificate of Incorporation
to be inapplicable or unenforceable in an action, we may incur additional costs associated with resolving such action in other jurisdictions,
which could harm our business, operating results and financial condition.
We
believe these provisions benefit us by providing increased consistency in the application of Delaware law by chancellors particularly
experienced in resolving corporate disputes, efficient administration of cases on a more expedited schedule relative to other forums
and protection against the burdens of multi-forum litigation. However, the provision may have the effect of discouraging lawsuits against
our directors, officers, employees, and agents as it may limit any stockholder’s ability to bring a claim in a judicial forum that
such stockholder finds favorable for disputes with us or our directors, officers, employees or agents. The enforceability of similar
choice of forum provisions in other companies’ certificates of incorporation has been challenged in legal proceedings, and it is
possible that, in connection with any applicable action brought against us, a court could find the choice of forum provisions contained
in our Certificate of Incorporation to be inapplicable or unenforceable in such action. If a court were to find the choice of forum provision
contained in our Certificate of Incorporation to be inapplicable or unenforceable in an action, we may incur additional costs associated
with resolving such action in other jurisdictions, which could adversely affect our business, financial condition or results of operations.
IN
ADDITION TO THE ABOVE RISKS, BUSINESSES ARE OFTEN SUBJECT TO RISKS NOT FORESEEN OR FULLY APPRECIATED BY MANAGEMENT. IN REVIEWING THIS
FILING, POTENTIAL INVESTORS SHOULD KEEP IN MIND THAT OTHER POSSIBLE RISKS MAY ADVERSELY IMPACT THE COMPANY’S BUSINESS OPERATIONS
AND THE VALUE OF THE COMPANY’S SECURITIES.
SPECIAL
NOTE REGARDING FORWARD-LOOKING STATEMENTS
This
prospectus contains “forward-looking statements.” Forward-looking statements reflect the current view about future events.
When used in this prospectus, the words “anticipate,” “believe,” “estimate,” “expect,”
“future,” “intend,” “plan” or the negative of these terms and similar expressions, as they relate
to us or our management, identify forward-looking statements. Such statements, include, but are not limited to, statements contained
in this prospectus relating to our business strategy, our future operating results and liquidity and capital resources outlook. Forward-looking
statements are based on our current expectations and assumptions regarding our business, the economy and other future conditions. Because
forward–looking statements relate to the future, they are subject to inherent uncertainties, risks and changes in circumstances
that are difficult to predict. Our actual results may differ materially from those contemplated by the forward-looking statements. They
are neither statements of historical fact nor guarantees of assurance of future performance. We caution you therefore against relying
on any of these forward-looking statements. Important factors that could cause actual results to differ materially from those in the
forward-looking statements include, without limitation:
| |
● |
Our ability to effectively
operate our business segments; |
| |
● |
Our ability to manage our
research, development, expansion, growth, and operating expenses; |
| |
● |
Our ability to evaluate
and measure our business, prospects, and performance metrics; |
| |
● |
Our ability to compete,
directly and indirectly, and succeed in the highly competitive medical devices industry; |
| |
● |
Our ability to respond
and adapt to changes in technology and customer behavior; |
| |
● |
Our ability to protect
our intellectual property and to develop, maintain and enhance a strong brand; and |
| |
● |
Other factors (including
the risks contained in the section of this prospectus entitled “Risk Factors”) relating to our industry, our operations
and results of operations. |
Should
one or more of these risks or uncertainties materialize, or should the underlying assumptions prove incorrect, actual results may differ
significantly from those anticipated, believed, estimated, expected, intended or planned.
Factors
or events that could cause our actual results to differ may emerge from time to time, and it is not possible for us to predict all of
them. We cannot guarantee future results, levels of activity, performance or achievements. Except as required by applicable law, including
the securities laws of the United States, we do not intend to update any of the forward-looking statements to conform these statements
to actual results.
USE
OF PROCEEDS
We
estimate that the net proceeds to us from this offering from the sale of the securities based on the assumed combined public offering
price of $[*] per share and accompanying Common Warrant, which represents the closing price of our common stock on [*], 2024 and assuming
no exercise of Pre-funded Warrants and Common Warrants will be approximately $[*] million, after deducting the Placement Agent fees and
estimated offering expenses.
The
principal purposes of this offering are to increase our capitalization and financial flexibility, increase our visibility in the marketplace
and create a public market for our common stock and the Warrants. As of the date of this prospectus, we cannot specify with certainty
all of the particular uses for the net proceeds to us from this offering. However, we currently intend to use the net proceeds from this
offering to hire additional employees, expand the commercial launch of our product including training clinicians on The CATAMARAN System
procedure, continuing clinical marketing studies that are focused on capturing post-market safety data, gathering system feedback and
initiating product refinements, other sales and marketing activities and for working capital and general corporate purposes. See “Business—Research
& Development.”
We
will retain broad discretion in the allocation of the net proceeds from this offering and could utilize the proceeds in ways that do
not necessarily improve our results of operations or enhance the value of our common stock.
The
table below sets forth the manner in which we expect to use the net proceeds we receive from this offering if the gross proceeds from
this public offering are 100% of the maximum offering amount. All amounts included in the table below are estimates.
| Description |
|
Total |
|
| Expand Catamaran commercialization
and physician training |
|
$ |
|
|
| Marketing, including clinical marketing studies |
|
$ |
|
|
| Additional hires |
|
$ |
|
|
| Working Capital and General Corporate Purposes |
|
$ |
|
|
| Total |
|
$ |
|
|
The
foregoing information is an estimate based on our current business plan. We may find it necessary or advisable to re-allocate portions
of the net proceeds reserved for one category to another, and we will have broad discretion in doing so. Pending these uses, we intend
to invest the net proceeds of this offering in a money market or other interest-bearing account. The
amounts and timing of our actual expenditures will depend upon numerous factors, including our sales and marketing and commercialization
efforts, demand for our products, our operating costs and the other factors described under “Risk Factors” in this
prospectus. Accordingly, our management will have flexibility in applying the net proceeds from this offering. An investor will not have
the opportunity to evaluate the economic, financial or other information on which we base our decisions on how to use the proceeds.
DIVIDEND
POLICY
We
have not declared any cash dividends since inception and we do not anticipate paying any dividends in the foreseeable future. Instead,
we anticipate that all of our earnings will be used to provide working capital, to support our operations, and to finance the growth
and development of our business, including potentially the acquisition of, or investment in, businesses, technologies or products that
complement our existing business. The payment of dividends is within the discretion of the board of directors and will depend on our
earnings, capital requirements, financial condition, prospects, applicable Delaware law, which provides that dividends are only payable
out of surplus or current net profits, and other factors our board might deem relevant. There are no restrictions that currently limit
our ability to pay dividends on our common stock other than those generally imposed by applicable state law.
CAPITALIZATION
The
following table sets forth our consolidated cash and capitalization, as of June 30, 2024. Such information is set forth on the following
basis:
| |
● |
on an as adjusted basis,
giving effect to the assumed sale by us of [*] shares of common stock in this offering at an assumed public offering price of $ [*]
per share (the last reported sale price of our common stock on Nasdaq on [*], 2024), after deducting the Placement Agent fees and
other estimated offering expenses payable by us and assuming no exercise of Pre-funded Warrants and Common Warrants. |
You
should read this table in conjunction with “Use of Proceeds” included in this prospectus and “Management’s
Discussion and Analysis of Financial Condition and Results of Operations,” and our financial statements for the period ended
June 30, 2024, and the related notes thereto included in this prospectus:
| | |
Actual | | |
As adjusted (1) | |
| Cash, cash equivalents and investments | |
$ | 1,968 | | |
| | |
| | |
| | | |
| | |
| Common stock, $0.001 par value; 130,000,000 shares authorized at June 30, 2024; 3,780,827 shares issued and outstanding at June 30, 2024; [*] shares issued and outstanding, as adjusted | |
$ | 4 | | |
| | |
| Additional paid-in capital | |
$ | 60,003 | | |
| | |
| Accumulated deficit | |
$ | (62,475 | ) | |
| | |
| Accumulated other comprehensive loss | |
$ | - | | |
| | |
| Total stockholders’ equity | |
$ | 832 | | |
| | |
| Total capitalization | |
$ | 2,800 | | |
| | |
| (1) | Does not include (i) 256,968 shares of our common stock underlying
our Series A Preferred Stock, which are convertible into 2,569,680 shares of common stock and vote with the common stock on an as converted
basis; and (ii) 45,000 shares of our common stock underlying our Note Warrants; (iii) 415,468 shares of our common stock underlying our
Series A Warrants; (iv) 209,253 shares of our common stock issuable pursuant to options and restricted stock units granted pursuant to
our equity incentive plan and (v) 9,600 shares our common stock issuable upon the exercise of the warrants issued to our underwriters
in our initial public offering. |
DILUTION
Purchasers
of our securities in this offering will experience an immediate and substantial dilution to the extent of the difference between the
public offering price per share of our common stock and the as adjusted net tangible book value per share of our common stock after this
offering.
The historical net tangible book value of our
common stock as of June 30, 2024 was $832,000 or $0.22 per share. Historical net tangible book value per share of our common stock represents
our total tangible assets (total assets less intangible assets) less total liabilities divided by the number of shares of our common stock
outstanding as of that date.
After
giving effect to the sale of shares of common stock in this offering at a public offering price of $[*] per share of common stock (the
last reported sale price of our common stock on Nasdaq on [*], 2024) and assuming no exercise of the Common Warrants and assuming no
sale of Pre-funded Warrants for net proceeds of approximately $[*] as if such offering and such share issuances had occurred on June
30, 2024, our as adjusted net tangible book value as June 30, 2024, would have been $[*] or approximately $[*] per share of our common
stock. This represents an immediate increase in net tangible book value per share of $[*] to the existing stockholders and an immediate
dilution in net tangible book value per share of $[*] to new investors (attributing no value to the Common Warrants and assuming no sale
of Pre-funded Warrants). We determine dilution by subtracting the as adjusted net tangible book value per share after this offering from
the amount of cash that a new investor paid for a share of common stock in this offering. The following table illustrates this per share
dilution to new investors:
| Public offering price per share | |
| | |
| |
| Historical net tangible book value per share as of June 30, 2024 | |
$ | 0.22 | | |
| | |
| Increase in net tangible book value per share after giving effect to the offering | |
$ | | | |
| | |
| As Adjusted net tangible book value per share as of June 30, 2024 | |
| | | |
| | |
| Dilution in net tangible book value per share to new investors | |
| | | |
| | |
The
dilution information discussed above is illustrative only and will change based on the actual public offering price and other terms of
this offering determined at pricing.
After
completion of this offering, our existing stockholders would own approximately [*]% and our new investors would own approximately [*]%
of the total number of shares of our common stock outstanding after this offering.
The above
discussion and table are based on 3,780,827 shares of our common stock outstanding as of
June 30, 2024, and excludes as of such date: (i) 256,968 shares of our common stock underlying our Series A Preferred Stock, which
are convertible into 2,625,016 shares of common stock and vote with the common stock on an as converted basis; and (ii) 45,000 shares
of our common stock underlying our Note Warrants; (iii) 415,468 shares of our common stock underlying our Series A Warrants; (iv) 209,253
shares of our common stock issuable pursuant to options and restricted stock units granted pursuant to our equity incentive plan and
(v) 9,600 shares our common stock issuable upon the exercise of the warrants issued to our underwriters in our initial public offering.
To
the extent that outstanding options or warrants are exercised, you will experience further dilution. In addition, we may choose to raise
additional capital due to market conditions or strategic considerations even if we believe we have sufficient funds for our current or
future operating plans. To the extent that additional capital is raised through the sale of equity or convertible debt securities, the
issuance of these securities may result in further dilution to our stockholders.
Capitalization
Table(1)
| | |
Shares Purchased(1) | | |
Total Consideration | | |
| |
| | |
Number
| | |
Percent | | |
Amount | | |
Percent | | |
Per Share | |
| Existing stockholders | |
| | | |
| | | |
| | | |
| | | |
| | |
| New Investors | |
| | | |
| | | |
| | | |
| | | |
| | |
| | |
| | | |
| | | |
| | | |
| | | |
| | |
| (1) | Assumes
no sale of Pre-funded Warrants. |
MARKET
FOR COMMON EQUITY AND RELATED STOCKHOLDER MATTERS
Our common stock and Tradeable Warrants are listed
on The Nasdaq Capital Market under the symbols “TNON” and “TNONW,” respectively. As of August 13, 2024, we have
3,951,767 shares of common stock issued and outstanding held by 63 stockholders of record.
We
also have outstanding:
| ● | 209,253
shares of our common stock issuable pursuant to options and restricted stock units granted pursuant to our equity incentive plan; |
| ● | 9,600
shares of our common stock issuable upon the exercise of warrants issued to the underwriters in our initial public offering that closed
on April 29, 2022; |
| ● | Warrants
to purchase up to 1,918,000 shares of our common stock at an exercise price equal to $3.146 per share issued to investors in our June
2023 public offering; |
| ● | Warrants
to purchase up to 45,000 shares of our common stock at an exercise price equal to $1.94 per share issued to investors in our November
2023 private placement; and |
| ● | Series A Warrants to purchase up to 415,468 shares of our common stock
at an exercise price equal to $1.2705. |
Securities
Authorized for Issuance under Equity Incentive Plan
In
January and February 2022, our Board and our shareholders approved our 2022 Equity Incentive Plan (the “2022 Plan,” together
with the 2012 Plan, the “Plans”). The 2022 Plan governs equity awards to our employees, directors, officers, consultants
and other eligible participants. Initially, the maximum number of shares of our common stock that may be subject to awards under the
2022 Plan is equal to (i) 160,000 plus (ii) the lesser of (a) 75,000 shares of our common stock and (b) the number of shares of our common
stock subject to awards granted under the 2012 Plan that after the 2012 Plan is terminated are canceled, expired or otherwise terminated
without having been exercised in full, are tendered to or withheld by the Company for payment of an exercise price or for tax withholding
obligations, or are forfeited to or repurchased by the Company due to failure to vest. The maximum number of shares that are subject
to awards under the 2022 Plan is subject to an annual increase equal to the lesser of (i) 110,000 shares of our common stock, (ii) a
number of shares of our common stock equal to 4% of the prior year’s maximum number and (iii) such number of shares of our common
stock as determined by the 2022 Plan administrator.
On July 23, 2024, at our annual meeting, our stockholders
voted to amend the 2022 Plan to increase the number of shares reserved for issuance under the 2022 Plan by 1,100,000 shares. There are
currently 1,408,959 shares reserved for issuance under the 2022 Plan, of which 1,154,173 shares are available for future grants. For a
more detailed description of the 2022 Plan see “Description of Securities-2022 Equity Incentive Plan.”
The
types of awards permitted under the Plans include nonqualified stock options, incentive stock options, stock appreciation rights, restricted
stock, restricted stock units, performance shares, performance units and other awards. Each option shall be exercisable at such times
and subject to such terms and conditions as the Board may specify.
The
Board has the power to amend, suspend or terminate the Plans without stockholder approval or ratification at any time or from time to
time. No change may be made that increases the total number of shares of our common stock reserved for issuance pursuant to incentive
awards or reduces the minimum exercise price for options or exchange of options for other incentive awards, unless such change is authorized
by our stockholders within one year.
On
April 8, 2024, we launched a one-time stock option exchange program (the “Option Exchange”) pursuant to which eligible participants
were able to exchange outstanding stock options for a lesser amount of new restricted stock units (“RSUs”). Our executive
officers, non-employee directors and consultants were eligible to participate in the Option Exchange. Employees, non-employee directors
and consultants received one RSU for every two shares of our common stock underlying the eligible options surrendered. This “exchange
ratio” (2-for-1) was applied on a grant-by-grant basis. The Option Exchange expired on May 6, 2024 at 11:59 p.m., Eastern Time.
At that time, stock options to purchase 83,391 shares of our common stock were surrendered and 41,698 new RSUs were issued under the
2022 Plan.
MANAGEMENT’S
DISCUSSION AND ANALYSIS OF
FINANCIAL
CONDITION AND RESULTS OF OPERATIONS
You
should read the following discussion and analysis of our financial condition and results of operations together with our financial statements
and the notes to those statements included elsewhere in this Registration Statement on Form S-1. In addition to historical financial
information, this discussion and analysis contains forward-looking statements that reflect our plans, estimates and beliefs. You should
not place undue reliance on these forward-looking statements, which involve risks and uncertainties. As a result of many factors, including
but not limited to those set forth under “Risk Factors,” our actual results may differ materially from those anticipated
in these forward-looking statements. See “Cautionary Note Regarding Forward-Looking Statements.”
Overview
Tenon
Medical, Inc., a medical device company formed in 2012, has developed a proprietary, U.S. Food and Drug Administration (“FDA”)
approved surgical implant-system, which we call The Catamaran™ SI Joint Fusion System (“The Catamaran System”). The
Catamaran System offers a novel, less invasive inferior-posterior approach to the sacroiliac joint (“SI Joint”) using a single,
robust titanium implant to treat SI Joint dysfunction that often causes severe lower back pain. The system features the Catamaran™
Fixation Device which passes through both the axial and sagittal planes of the ilium and sacrum, transfixing the SI Joint along its longitudinal
axis. Published clinical studies have shown that 15% to 30% of all chronic lower back pain is associated with the SI Joint.
With
an entry similar to the SI Joint injection, the surgical approach is direct to the joint. The angle and trajectory of the inferior-posterior
approach is designed to point away from critical neural and vascular structures and into the strongest cortical bone. Joined by a patented
osteotome bridge, the implant design consists of two hollow fenestrated pontoons with an open framework to facilitate bony in-growth
through the SI Joint. One pontoon fixates into the ilium and the other into the sacrum. The osteotome is designed to disrupt the articular
portion of the joint to help facilitate a fusion response.
Our
initial clinical results indicate that the Catamaran System implant is promoting fusion across the joint as evidenced by computerized
tomography (CT) scans which is the gold standard widely accepted by the clinical community. We had our national launch of The Catamaran
System in October 2022 and are building a sales and marketing infrastructure to market our product and address the greatly underserved
market opportunity that exists.
We
believe that the implant design and procedure we have developed, along with the 2D and 3D protocols for proper implantation will be received
well by the clinician community who have been looking for a next generation device.
We
have incurred net losses since our inception in 2012. As of June 30, 2024, we had an accumulated deficit of approximately $62.5 million.
To date, we have financed our operations primarily through an initial public offering, private placements of equity securities, certain
debt-related financing arrangements, and sales of our product. We have devoted substantially all of our resources to research and development,
regulatory matters and sales and marketing of our product.
2024
Series A Offering
On
February 20, 2024, we entered into a Securities Purchase Agreement (the “Series A Purchase Agreement”) with certain investors
(the “Series A Investors”), pursuant to which the Company agreed to sell, issue and deliver to the Series A Investors, in
a private placement offering (the “Series A Offering”), a total of 172,239 shares of the Company’s Series A Preferred
Stock (the “Series A Preferred Stock”) and warrants (the "Warrants”) to purchase 258,374 shares of our common
stock, par value $0.001 per share, at an exercise price equal to $1.2705 per share for an aggregate offering price of $2,605,000. Under
the Series A Purchase Agreement, each Series A Investor paid $15.125 for each share of Series A Preferred Stock and along with their
shares of Series A Preferred Stock, received Warrants equal to 15% of the number of shares of our common stock initially underlying such
shares of Series A Preferred Stock. In connection with the offering of the Series A Preferred Stock the Company exchanged the Notes (as
defined below) for 84,729 shares of Series A Preferred Stock and Series A Warrants to purchase 157,094 shares of our common stock. There
are a total of 256,968 shares of Series A Preferred Stock outstanding as of June 30, 2024.
Reverse
Stock Split
On
November 2, 2023, the Company effected a 1-for-10 reverse stock split by filing an amendment to the Company’s Amended and Restated
Certificate of Incorporation, as amended, with the Delaware Secretary of State. The reverse stock split combined every ten shares of
our common stock issued and outstanding immediately prior to effecting the reverse stock split into one share of common stock. No fractional
shares were issued in connection with the reverse stock split. All historical share and per share amounts reflected throughout this document
have been adjusted to reflect the reverse stock split. The authorized number of shares and the par value per share of the Company’s
common stock were not affected by the reverse stock split.
Components
of Results of Operations
Revenue
We
derive substantially all our revenue from sales of The Catamaran System to a limited number of clinicians. Revenue from sales of The
Catamaran System fluctuates based on volume of cases (procedures performed), discounts, and the number of implants used for a particular
patient. Similar to other orthopedic companies, our revenue can also fluctuate from quarter to quarter due to a variety of factors, including
reimbursement, changes in independent sales representatives and physician activities.
Cost
of Goods Sold, Gross Profit, and Gross Margin
We
utilize contract manufacturers for production of The Catamaran System implants and Catamaran Tray Sets. Cost of goods sold consists primarily
of overhead related to operation personnel and facility costs, costs of the components of The Catamaran System implants and instruments,
quality inspection, packaging, scrap and inventory obsolescence, as well as distribution-related expenses such as logistics and shipping
costs. We anticipate that certain of our cost of goods sold will increase in absolute dollars as case levels increase.
Our
gross margins have been and will continue to be affected by a variety of factors, including the cost to have our product manufactured
for us, pricing pressure from increasing competition, and the factors described above impacting our revenue.
Operating
Expenses
Our
operating expenses consist of sales and marketing, research and development, and general and administrative expenses. Personnel costs
are the most significant component of operating expenses and consist of consulting expenses, salaries, sales commissions and other cash
and stock-based compensation related expenses. We expect operating expenses to increase in absolute dollars as we continue to invest
and grow our business.
Sales
and Marketing Expenses
Sales
and marketing expenses primarily consist of salaries, commissions, stock-based compensation expense and travel and entertainment expenses
of our sales and market personnel along with commissions paid to our independent distributors. We expect our sales and marketing expenses
to increase in absolute dollars with the increased sales of The Catamaran System resulting in higher commissions and salaries, increased
clinician and sales representative training, and the cost to complete our clinical study to gain wider clinician adoption of The Catamaran
System. Our sales and marketing expenses may fluctuate from period to period due to timing of sales and marketing activities related
to the commercial activity of our product.
Research
and Development Expenses
Our
research and development expenses primarily consist of engineering, product development, regulatory expenses, and consulting services,
outside prototyping services, outside research activities, materials, and other costs associated with the development and refinement
of our product. Research and development expenses also include related personnel and consultants’ compensation and stock-based
compensation expense. We expense research and development costs as they are incurred. We expect research and development expense to increase
in absolute dollars as we improve The Catamaran System, develop new products, add research and development personnel, and undergo clinical
activities that may be required for regulatory clearances of future products.
General
and Administrative Expenses
General
and administrative expenses primarily consist of salaries, consultants’ compensation, stock-based compensation expense, and other
costs for finance, accounting, legal, compliance, and administrative matters. We expect our general and administrative expenses to increase
in absolute dollars as we add personnel and information technology infrastructure to support the growth of our business. We also expect
to incur additional general and administrative expenses as a result of operating as a public company, including but not limited to: expenses
related to compliance with the rules and regulations of the Securities and Exchange Commission and those of The Nasdaq Stock Market LLC
on which our securities are traded; additional insurance expenses; investor relations activities; and other administrative and professional
services. While we expect the general and administrative expenses to increase in absolute dollars, we anticipate that it will decrease
as a percentage of revenue over time.
Gain
on Investments, Interest Expense and Other Income (Expense), Net
Gain
on investments consists of interest income and realized gains and losses from the sale of our investments in money market and corporate
debt securities. Interest expense is related to borrowings. Other income and expenses have not been significant to date.
Results
of Operations
The
following table sets forth our results of operations for the periods presented (in thousands):
| | |
Three Months Ended June 30, | | |
Six Months Ended
June 30, | |
| Consolidated Statements of Operations Data: | |
2024 | | |
2023 | | |
2024 | | |
2023 | |
| Revenue | |
$ | 901 | | |
$ | 743 | | |
$ | 1,620 | | |
$ | 1,176 | |
| Cost of goods sold | |
| 431 | | |
| 549 | | |
| 680 | | |
| 1,029 | |
| Gross (loss) profit | |
| 470 | | |
| 194 | | |
| 940 | | |
| 147 | |
| Operating expenses: | |
| | | |
| | | |
| | | |
| | |
| Research and development | |
| 708 | | |
| 901 | | |
| 1,377 | | |
| 1,735 | |
| Sales and marketing | |
| 1,448 | | |
| 1,883 | | |
| 2,829 | | |
| 3,909 | |
| General and administrative | |
| 2,186 | | |
| 1,732 | | |
| 4,112 | | |
| 3,711 | |
| Total operating expenses | |
| 4,342 | | |
| 4,516 | | |
| 8,318 | | |
| 9,355 | |
| Loss from operations | |
| (3,872 | ) | |
| (4,322 | ) | |
| (7,378 | ) | |
| (9,208 | ) |
| Interest and other income (expense), net: | |
| | | |
| | | |
| | | |
| | |
| Gain on investments | |
| 39 | | |
| 37 | | |
| 66 | | |
| 93 | |
| Interest expense | |
| — | | |
| — | | |
| (34 | ) | |
| — | |
| Other income (expense) | |
| 7 | | |
| — | | |
| (56 | ) | |
| — | |
| Net loss | |
$ | (3,826 | ) | |
$ | (4,285 | ) | |
$ | (7,402 | ) | |
$ | (9,115 | ) |
| | |
Years Ended December 31, | |
| Consolidated Statements of Operations Data in Dollars: | |
2023 | | |
2022 | |
| Revenue | |
$ | 2,928 | | |
$ | 691 | |
| Cost of goods sold | |
| 1,687 | | |
| 1,332 | |
| Gross profit (loss) | |
| 1,241 | | |
| (641 | ) |
| Operating expenses: | |
| | | |
| | |
| Research and development | |
| 3,163 | | |
| 2,828 | |
| Sales and marketing | |
| 6,778 | | |
| 7,833 | |
| General and administrative | |
| 7,027 | | |
| 7,423 | |
| Total operating expenses | |
| 16,968 | | |
| 18,084 | |
| Loss from operations | |
| (15,727 | ) | |
| (18,725 | ) |
| Interest and other income (expense), net: | |
| | | |
| | |
| Gain on investments | |
| 167 | | |
| 180 | |
| Interest expense | |
| (21 | ) | |
| (354 | ) |
| Other expense | |
| — | | |
| (18 | ) |
| Net loss | |
$ | (15,581 | ) | |
$ | (18,917 | ) |
The
following table sets forth our results of operations as a percentage of revenue:
| | |
Three Months Ended
June 30, | | |
Six Months Ended
June 30, | |
| Consolidated Statements of Operations Data: | |
2024 | | |
2023 | | |
2024 | | |
2023 | |
| Revenue | |
| 100 | % | |
| 100 | % | |
| 100 | % | |
| 100 | % |
| Cost of goods sold | |
| 48 | | |
| 74 | | |
| 42 | | |
| 88 | |
| Gross profit | |
| 52 | | |
| 26 | | |
| 58 | | |
| 12 | |
| Operating expenses: | |
| | | |
| | | |
| | | |
| | |
| Research and development | |
| 79 | | |
| 121 | | |
| 85 | | |
| 148 | |
| Sales and marketing | |
| 161 | | |
| 253 | | |
| 175 | | |
| 332 | |
| General and administrative | |
| 243 | | |
| 233 | | |
| 254 | | |
| 316 | |
| Total operating expenses | |
| 482 | | |
| 608 | | |
| 513 | | |
| 795 | |
| Loss from operations | |
| (430 | ) | |
| (582 | ) | |
| (455 | ) | |
| (783 | ) |
| Interest and other income (expense), net: | |
| | | |
| | | |
| | | |
| | |
| Gain on investments | |
| 4 | | |
| 5 | | |
| 4 | | |
| 8 | |
| Interest expense | |
| — | | |
| — | | |
| (2 | ) | |
| — | |
| Other expense | |
| 1 | | |
| — | | |
| (3 | ) | |
| — | |
| Net loss | |
| (425 | )% | |
| (577 | )% | |
| (457 | )% | |
| (775 | )% |
| | |
Years Ended December 31, | |
| Consolidated Statements of Operations Data as a Percent of Revenue: | |
2023 | | |
2022 | |
| Revenue | |
| 100 | % | |
| 100 | % |
| Cost of goods sold | |
| 58 | | |
| 193 | |
| Gross profit (loss) | |
| 42 | | |
| (93 | ) |
| Operating expenses: | |
| | | |
| | |
| Research and development | |
| 108 | | |
| 409 | |
| Sales and marketing | |
| 231 | | |
| 1,134 | |
| General and administrative | |
| 240 | | |
| 1,074 | |
| Total operating expenses | |
| 580 | | |
| 2,617 | |
| Loss from operations | |
| (537 | ) | |
| (2,710 | ) |
| Interest and other income (expense), net: | |
| | | |
| | |
| Gain on investments | |
| 6 | | |
| 26 | |
| Interest expense | |
| (1 | ) | |
| (51 | ) |
| Other expense | |
| — | | |
| (3 | ) |
| Net loss | |
| (532 | )% | |
| (2,738 | )% |
Comparison
of the Three and Six Months Ended June 30, 2024 and 2023 (in thousands, except percentages)
Revenue,
Cost of Goods Sold, Gross Profit, and Gross Margin
| | |
Three Months Ended
June 30, | | |
| | |
| |
| | |
2024 | | |
2023 | | |
$ Change | | |
% Change | |
| Revenue | |
$ | 901 | | |
$ | 743 | | |
$ | 158 | | |
| 21 | % |
| Cost of goods sold | |
| 431 | | |
| 549 | | |
| (118 | ) | |
| (21 | )% |
| Gross profit | |
$ | 470 | | |
$ | 194 | | |
$ | 276 | | |
| 142 | % |
| Gross profit percentage | |
| 52 | % | |
| 26 | % | |
| | | |
| | |
| | |
Six Months Ended
June 30, | | |
| | |
| |
| | |
2024 | | |
2023 | | |
$ Change | | |
% Change | |
| Revenue | |
$ | 1,620 | | |
$ | 1,176 | | |
$ | 444 | | |
| 38 | % |
| Cost of goods sold | |
| 680 | | |
| 1,029 | | |
| (349 | ) | |
| (34 | )% |
| Gross profit | |
$ | 940 | | |
$ | 147 | | |
$ | 793 | | |
| 539 | % |
| Gross profit percentage | |
| 58 | % | |
| 13 | % | |
| | | |
| | |
Revenue. The
increase in revenue for the three and six months ended June 30, 2024 as compared to the same periods in 2023 was primarily due to increases
of 7% and 19%, respectively, in the number of surgical procedures in which the Catamaran System was used.
Cost of Goods Sold, Gross Profit, and Gross
Margin. The change in cost of goods sold for the three and six months ended June 30, 2024 as compared to the same periods in
2023 was due to increases of 7% and 19%, respectively, in the number of surgical procedures performed. Gross profit and gross margin percentage
for the three and six month periods ended June 30, 2024 as compared to the same periods in 2023 improved due to higher revenue associated
with the increase in the number of surgical procedures, operating leverage created due to lower relative fixed costs and the absorption
of more production overhead costs into our standard cost.
Operating
Expenses
| | |
Three Months Ended
June 30, | | |
| | |
| |
| | |
2024 | | |
2023 | | |
$ Change | | |
% Change | |
| Research and development | |
$ | 708 | | |
$ | 901 | | |
$ | (193 | ) | |
| (21 | )% |
| Sales and marketing | |
| 1,448 | | |
| 1,883 | | |
| (435 | ) | |
| (23 | )% |
| General and administrative | |
| 2,186 | | |
| 1,732 | | |
| 454 | | |
| 26 | % |
| Total operating expenses | |
$ | 4,342 | | |
$ | 4,516 | | |
$ | (174 | ) | |
| (4 | )% |
| | |
Six Months Ended
June 30, | | |
| | |
| |
| | |
2024 | | |
2023 | | |
$ Change | | |
% Change | |
| Research and development | |
$ | 1,377 | | |
$ | 1,735 | | |
$ | (358 | ) | |
| (21 | )% |
| Sales and marketing | |
| 2,829 | | |
| 3,909 | | |
| (1,080 | ) | |
| (28 | )% |
| General and administrative | |
| 4,112 | | |
| 3,711 | | |
| 401 | | |
| 11 | % |
| Total operating expenses | |
$ | 8,318 | | |
$ | 9,355 | | |
$ | (1,037 | ) | |
| (11 | )% |
Research
and Development Expenses. Research and development expenses for the three months ended June 30, 2024 decreased as compared to the
same period in 2023 primarily due to decreased professional fees ($141), payroll expenses ($29) and stock-based compensation ($19). Research
and development expenses for the six months ended June 30, 2024 decreased as compared to the same period in 2023 primarily due to decreased
professional fees ($279), payroll expenses ($111) and stock-based compensation ($25).
Sales
and Marketing Expenses. Sales and marketing expenses for the three months ended June 30, 2024 decreased as compared to the same
period in 2023 primarily due to SpineSource transition fees in 2023 ($260), decreased payroll and employee expenses ($165) and consulting
and professional fees ($102) partially offset by increased commission expense ($108). Sales and marketing expenses for the six months
ended June 30, 2024 decreased as compared to the same period in 2023 primarily due to SpineSource transition fees in 2023 ($690), decreased
payroll and employee expenses ($312) and consulting and professional fees ($197) partially offset by increased commission expense ($153).
General
and Administrative Expenses. General and administrative expenses for the three months ended June 30, 2024 increased as compared to
the same period in 2023 primarily due to increased legal and professional service fees ($215), insurance costs ($93), bad debt expense
($36) and stock-based compensation ($25). General and administrative expenses for the six months ended June 30, 2024 increased as compared
to the same period in 2023 primarily due to increased insurance costs ($191), legal and professional service fees ($93), bad debt expense
($46) and stock-based compensation ($22).
Gain
on Investments, Interest Expense and Other Income (Expense), Net
Gain
on investments for the six months ended June 30, 2024 decreased approximately $28 as compared to the six months ended June 30, 2023 due
to interest on our investments in money market and corporate debt securities. We did not have significant investments in corporate debt
securities during the first six months of 2024. Interest expense for the six months ended June 30, 2024 related to our convertible debt.
Other expense, net was related to foreign exchange losses on the liquidation of our Swiss subsidiary.
Liquidity
and Capital Resources
As
of June 30, 2024, we had cash and cash equivalents of $2.0 million. Since inception, we have financed our operations through private
placements of preferred stock, debt financing arrangements, our initial public offering, additional stock offerings and the sale of our
products. As of June 30, 2024, we had no outstanding debt.
As
of June 30, 2024, we had an accumulated deficit of $62.5 million and expect to incur additional losses in the future. We have not
achieved positive cash flow from operations to date. Based upon our current operating plan, our existing cash and cash equivalents will
not be sufficient to fund our operating expenses and working capital requirements through at least the next 12 months from the date these
consolidated financial statements were available to be released. We plan to raise the necessary additional capital through one or a combination
of public or private equity offerings, debt financings, and collaborations. We continue to face challenges and uncertainties and, as
a result, our available capital resources may be consumed more rapidly than currently expected due to (a) the uncertainty of future revenues
from The Catamaran System; (b) changes we may make to the business that affect ongoing operating expenses; (c) changes we may make in
our business strategy; (d) regulatory developments affecting our existing products; (e) changes we may make in our research and development
spending plans; and (f) other items affecting our forecasted level of expenditures and use of cash resources.
On
February 20, 2024, we entered into the Series A Purchase Agreement with certain investors, pursuant to which we agreed to sell, issue
and deliver to these investors, in a private placement offering, a total of 172,239 shares of our Series A Preferred Stock and warrants
to purchase 258,374 shares of our common stock, par value $0.001 per share, at an exercise price equal to $1.2705 per share for an aggregate
offering price of $2,605,000. Additionally, on February 20, 2024, the Series A Investors agreed to a complete prepayment of our obligations
under convertible notes (the “Convertible Notes”), including accrued interest, in exchange for 84,729 shares of Series A
Preferred Stock and warrants to purchase 157,094 shares of our common stock at $1.2705 per share and the Convertible Notes were cancelled.
The Series A Warrants are immediately exercisable and expire five years from the date of issuance. There are a total of 256,968 shares
of Series A Preferred Stock outstanding as of May 14, 2024.
As
we attempt to raise additional capital to fund our operations, funding may not be available to us on acceptable terms, or at all. If
we are unable to obtain adequate financing when needed, we may have to delay, reduce the scope of or suspend one or more of our sales
and marketing efforts, research and development activities, or other operations. We may seek to raise any necessary additional capital
through a combination of public or private equity offerings, debt financings, and collaborations. If we do raise additional capital through
public or private equity offerings, the ownership interest of our existing stockholders will be diluted, and the terms of these securities
may include liquidation or other preferences that adversely affect our stockholders’ rights. If we raise additional capital through
debt financing, we may be subject to covenants limiting or restricting our ability to take specific actions, such as incurring additional
debt, making capital expenditures, or declaring dividends. If we are unable to raise capital, we will need to delay, reduce, or terminate
planned activities to reduce costs. Doing so will likely harm our ability to execute our business plans. Due to the uncertainty in our
ability to raise capital, management believes that there is substantial doubt in our ability to continue as a going concern for the next
twelve months from the issuance of these consolidated financial statements.
Cash
Flows (in thousands, except percentages)
The
following table sets forth the primary sources and uses of cash for each of the periods presented below:
| | |
Six Months Ended
June 30, | | |
| | |
| |
| | |
2024 | | |
2023 | | |
$ Change | | |
% Change | |
| Net cash (used in) provided by: | |
| | |
| | |
| | |
| |
| Operating activities | |
$ | (4,758 | ) | |
$ | (6,752 | ) | |
$ | 1,994 | | |
| (30 | )% |
| Investing activities | |
| (119 | ) | |
| 5,798 | | |
| (5,917 | ) | |
| (102 | )% |
| Financing activities | |
| 4,371 | | |
| 4,665 | | |
| (294 | ) | |
| (6 | )% |
| Effect of foreign currency translation on cash flow | |
| 46 | | |
| 12 | | |
| 34 | | |
| 283 | % |
| Net (decrease) increase in cash and cash equivalents | |
$ | (460 | ) | |
$ | 3,723 | | |
$ | (4,183 | ) | |
| (112 | )% |
The
decrease in net cash used in operating activities for the six months ended June 30, 2024 as compared to the six months ended June 30,
2023 was primarily attributable to our decreased net loss of $1.7 million, adjusted for decreases in non-cash stock-based compensation
expenses ($42), in addition to increased accounts payable ($316), partially offset by decreases in accrued expenses ($313).
Cash
provided by investing activities for the six months ended June 30, 2024 consisted primarily purchases of property and equipment ($119).
Cash provided by investing activities for the six months ended June 30, 2023 consisted primarily of the net sales of short-term investments
($6,010) to fund our operations, partially offset by purchases of property and equipment ($212).
Cash
provided by financing activities for the six months ended June 30, 2024 consisted primarily of proceeds from the issuance of Series A
Convertible Preferred Stock ($2,437) and from the issuance of common stock ($1,934). Cash provided by financing activities for the six
months ended June 30, 2023 consisted primarily of the $4.8 million, net of relevant expenses, received from our registered offering in
June 2023.
Critical
Accounting Policies, Significant Judgments, and Use of Estimates
Our
management’s discussion and analysis of our financial condition and results of operations is based on our financial statements,
which have been prepared in accordance with U.S. GAAP. The preparation of these financial statements requires us to make estimates and
assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the
date of the financial statements, as well as the reported results of operations during the reporting periods. Our estimates are based
on our historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which
form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from three other
sources. Actual results could differ from these estimates under different assumptions or conditions. For the six months ended June 30,
2024, there were no significant changes to our existing critical accounting policies from those disclosed on our Annual Report on Form
10-K.
Off-Balance Sheet
Arrangements
As
of June 30, 2024, and December 31, 2023, we did not have any relationships with unconsolidated organizations or financial partnerships,
such as structured finance or special purpose entities that would have been established for the purpose of facilitating off-balance sheet
arrangements or other contractually narrow or limited purposes.
Comparison
of the years ended December 31, 2023 and 2022 (in thousands, except percentages)
Revenue,
Cost of Goods Sold, Gross Profit, and Gross Margin
| | |
Years Ended
December 31, | | |
| | |
| |
| | |
2023 | | |
2022 | | |
$ Change | | |
% Change | |
| Revenue | |
$ | 2,928 | | |
$ | 691 | | |
$ | 2,237 | | |
| 324 | % |
| Cost of goods sold | |
| 1,687 | | |
| 1,332 | | |
| 355 | | |
| 27 | % |
| Gross profit (loss) | |
$ | 1,241 | | |
$ | (641 | ) | |
$ | 1,882 | | |
| (294 | )% |
| Gross profit (loss) percentage | |
| 42 | % | |
| (93 | )% | |
| | | |
| | |
Revenue. The
increase in revenue for the year ended December 31, 2023 as compared to 2022 was primarily due to an increase of 312% in the number of
surgical procedures in which the Catamaran System was used.
Cost
of Goods Sold, Gross Profit, and Gross Margin. The increase in cost of goods sold for the year ended December 31, 2023 as compared
to 2022 was due to an increase of 312% in the number of surgical procedures performed. Gross profit (loss) and gross margin percentage
improved due to higher revenue associated with the increase in the number of surgical procedures, operating leverage created due to lower
relative fixed costs and the absorption of more overhead into our standard cost.
Operating
Expenses
| | |
Years Ended
December 31, | | |
| | |
| |
| | |
2023 | | |
2022 | | |
$ Change | | |
% Change | |
| Research and development | |
$ | 3,163 | | |
$ | 2,828 | | |
$ | 335 | | |
| 12 | % |
| Sales and marketing | |
| 6,778 | | |
| 7,833 | | |
| (1,055 | ) | |
| (13 | )% |
| General and administrative | |
| 7,027 | | |
| 7,423 | | |
| (396 | ) | |
| (5 | )% |
| Total operating expenses | |
$ | 16,968 | | |
$ | 18,084 | | |
$ | (1,116 | ) | |
| | |
Research
and Development Expenses. Research and development expenses for the year ended December 31, 2023 increased as compared to 2022
primarily due to increased stock-based compensation ($509) and payroll expenses ($49), partially offset by decreased professional fees
($137).
Sales
and Marketing Expenses. Sales and marketing expenses for the year ended December 31, 2023 decreased as compared to 2022 primarily
due to payments in 2022 in association with the termination of the SpineSource sales agreement ($3,611) and decreased consulting and
professional fees ($1,190), partially offset by increased payroll expenses ($2,388), sales commissions ($1,388) and stock-based compensation
($100). The increase in payroll and payroll related expenses is primarily due to the increased number of sales and marketing employees
as we build out our sales function.
General
and Administrative Expenses. General and administrative expenses for the year ended December 31, 2023 decreased as compared to 2022
primarily due to a legal settlement accrual in 2022 ($574) and decreased professional service fees ($852), partially offset by increased
stock-based compensation ($639) and payroll expenses ($271).
Gain
(Loss) on Investments, Interest Expense and Other Income (Expense), Net
| | |
Years Ended
December 31, | | |
| | |
| |
| | |
2023 | | |
2022 | | |
$ Change | | |
% Change | |
| Gain on investments | |
$ | 167 | | |
$ | 180 | | |
$ | (13 | ) | |
| 7 | % |
| Interest expense | |
| (21 | ) | |
| (354 | ) | |
| 333 | | |
| (94 | )% |
| Other expense, net | |
| — | | |
| (18 | ) | |
| 18 | | |
| 100 | % |
| Total operating expenses | |
$ | 146 | | |
$ | (192 | ) | |
$ | 338 | | |
| | |
Gain
on Investments. Gain on investments for the year ended December 31, 2023 decreased as compared to 2022 due to interest on our lower
amounts of investments in money market and corporate debt securities.
Interest
Expense. Interest expense for the year ended December 31, 2023 decreased as compared to 2022 primarily due to the conversion of our
convertible debt in association with our initial public offering in April 2022.
Other
Expense, Net. Other income and expenses were not significant during the twelve months ended December 31, 2023 and 2022.
Liquidity
and Capital Resources
As
of December 31, 2023, we had cash and cash equivalents of $2.4 million. Since inception, we have financed our operations through
private placements of preferred stock, debt financing arrangements, our initial public offering and the sale of our products. As of December
31, 2023, we had outstanding debt of $1.2 million.
As
of December 31, 2023, we had an accumulated deficit of $55.1 million. During the years ended December 31, 2023 and 2022, we incurred
net losses of $15.6 million and $18.9 million, respectively, and expect to incur additional losses in the future. We have not achieved
positive cash flow from operations to date. Based upon our current operating plan, our existing cash and cash equivalents will not be
sufficient to fund our operating expenses and working capital requirements through at least the next 12 months from the date these consolidated
financial statements were available to be released. We plan to raise the necessary additional capital through one or a combination of
public or private equity offerings, debt financings, and collaborations. We continue to face challenges and uncertainties and, as a result,
our available capital resources may be consumed more rapidly than currently expected due to (a) the uncertainty of future revenues from
The Catamaran System; (b) changes we may make to the business that affect ongoing operating expenses; (c) changes we may make in our
business strategy; (d) regulatory developments affecting our existing products; (e) changes we may make in our research and development
spending plans; and (f) other items affecting our forecasted level of expenditures and use of cash resources.
On
February 20, 2024, we entered into a Securities Purchase Agreement with certain investors, pursuant to which we agreed to sell, issue
and deliver to these investors, in a private placement offering, a total of 172,239 shares of our Series A Preferred Stock and warrants
to purchase 258,374 shares of our common stock, par value $0.001 per share, at an exercise price equal to $1.2705 per share for an aggregate
offering price of $2,605,000.
As
we attempt to raise additional capital to fund our operations, funding may not be available to us on acceptable terms, or at all. If
we are unable to obtain adequate financing when needed, we may have to delay, reduce the scope of or suspend one or more of our sales
and marketing efforts, research and development activities, or other operations. We may seek to raise any necessary additional capital
through a combination of public or private equity offerings, debt financings, and collaborations. If we do raise additional capital through
public or private equity offerings, the ownership interest of our existing stockholders will be diluted, and the terms of these securities
may include liquidation or other preferences that adversely affect our stockholders’ rights. If we raise additional capital through
debt financing, we may be subject to covenants limiting or restricting our ability to take specific actions, such as incurring additional
debt, making capital expenditures, or declaring dividends. If we are unable to raise capital, we will need to delay, reduce, or terminate
planned activities to reduce costs. Doing so will likely harm our ability to execute our business plans. Due to the uncertainty in our
ability to raise capital, management believes that there is substantial doubt in our ability to continue as a going concern for the next
twelve months from the issuance of these consolidated financial statements.
Contractual
Obligations
The
following table summarizes our contractual obligations as of December 31, 2023:
| | |
Payments Due By Period (In thousands) | |
| | |
| | |
Less than | | |
| | |
| | |
More
than | |
| | |
Total | | |
1 year | | |
1-3 years | | |
4-5 years | | |
5 years | |
| Operating leases | |
$ | 756 | | |
$ | 302 | | |
$ | 454 | | |
$ | — | | |
$ | — | |
| Convertible debt (1) | |
| 1,260 | | |
| 1,260 | | |
| — | | |
| — | | |
| — | |
| Total | |
$ | 2,016 | | |
$ | 1,562 | | |
$ | 454 | | |
$ | — | | |
$ | — | |
| (1) | Amount
represents the principal and accrued interest on the convertible debt as of December 31,
2023. Per the terms of the convertible debt, the entire amount was converted to preferred
stock in February 2024. |
Obligations
under Terminated Sales Representative Agreement: On October 6, 2022, we entered into the Terminating Amended and Restated Exclusive
Sales Representative Agreement (the “Termination Agreement”). In accordance with the Termination Agreement, (i) we paid the
Representative $1,000 in cash; and (ii) we agreed to pay the Representative (a) $85 per month during the six months after the date of
the Termination Agreement in return for efforts by the Representative to transition operations to us, (b) 20% of net sales of the Product
sold in the United States and Puerto Rico until December 31, 2023 and (c) after December 31, 2023, 10% of net sales until such time as
the aggregate amount paid to the Representative under this clause (c) and clause (b) above equal $3,600. In the event of an acquisition,
we will pay the Representative $3,600 less previous amounts paid pursuant to clause (b) and clause (c) above. The timing of the payments
under clause (b) and (c) is variable depending on the timing of our sales.
Cash Flows (in thousands, except percentages)
The
following table sets forth the primary sources and uses of cash for each of the periods presented below:
| | |
Years Ended
December 31, | | |
| | |
| |
| | |
2023 | | |
2022 | | |
$ Change | | |
% Change | |
| Net cash (used in) provided by: | |
| | |
| | |
| | |
| |
| Operating activities | |
$ | (12,183 | ) | |
$ | (12,025 | ) | |
$ | (158 | ) | |
| 1 | % |
| Investing activities | |
| 6,142 | | |
| (2,884 | ) | |
| 9,026 | | |
| (313 | )% |
| Financing activities | |
| 6,302 | | |
| 14,114 | | |
| (7,812 | ) | |
| (55 | )% |
| Effect of foreign currency translation on cash flow | |
| 38 | | |
| 7 | | |
| 31 | | |
| (443 | )% |
| Net increase (decrease) in cash and cash equivalents | |
$ | 299 | | |
$ | (788 | ) | |
$ | 1,087 | | |
| (138 | )% |
The
increase in net cash used in operating activities for the year ended December 31, 2023 as compared to 2022 was primarily attributable
to decreases in our accrued expenses ($2,387) and accounts payable ($189) and increases in prepaid expenses ($244) and accounts receivable
($138), partially offset by our decreased net loss ($3,336), adjusted for increases in non-cash stock-based compensation expenses ($1,248)
and a decrease in common stock issued for services ($1,561).
Cash
provided by investing activities for the year ended December 31, 2023 consisted primarily of the net sales of short-term investments
of approximately $6.5 million as used those amounts to fund operations, partially offset by purchases of property and equipment of $0.4
million as we acquired the components for our surgical tray sets. Cash used in investing activities for the year ended December 31, 2022
consisted primarily of the net purchase of short-term investments of approximately $2.0 million as we invested a portion of our IPO proceeds,
in addition to purchases of property and equipment of $0.8 million as we acquired the components for our surgical tray sets.
Cash
provided by financing activities for the year ended December 31, 2023 consisted of the $5.3 million, net of relevant expenses, received
from our offerings of stock in 2023 in addition to $1.2 million from the issuance of the Convertible Notes. Cash provided by financing
activities for the year ended December 31, 2022 consisted of the $14.1 million cash received from our initial public offering in April
2022, net of relevant expenses.
Off-Balance Sheet
Arrangements
As
of December 31, 2023 and 2022, we did not have any relationships with unconsolidated organizations or financial partnerships, such as
structured finance or special purpose entities that would have been established for the purpose of facilitating off-balance sheet
arrangements or other contractually narrow or limited purposes.
BUSINESS
Introduction
Tenon
Medical, Inc. was incorporated in the State of Delaware on June 19, 2012 and was headquartered in San Ramon, California until June 2021
when it relocated to Los Gatos, California. The Company is a medical device company that has developed The Catamaran™ SI Joint
Fusion System that offers a novel, less invasive approach to the sacroiliac joint (the “SI Joint”) using a single, robust,
titanium implant for treatment of the most common types of SI Joint disorders that cause lower back pain. The Company received U.S. Food
and Drug Administration (“FDA”) clearance in 2018 for The Catamaran System and is currently focused on the US market. Since
the national launch of The Catamaran System in October 2022, the Company is focused on three commercial opportunities: 1) Primary SI
Joint procedures, 2) Revision procedures of failed SI Joint implants and 3) SI Joint fusion adjunct to a spine fusion construct.
The
Opportunity
We
estimate that over 30 million American adults have chronic lower back pain. Published clinical studies have shown that 15% to 30% of
all chronic lower back pain is associated with the SI-Joint. For patients whose chronic lower back pain stems from the Sacroiliac Joint
(“SI-Joint”), our experience in both clinical trials and commercial settings indicates the system to be introduced by Tenon
could be beneficial for patients who are properly diagnosed and screened for surgery by trained healthcare providers.
In
2019, approximately 475,000 patients in the United States were estimated to have received an aesthetic injection to temporarily alleviate
pain emanating from the SI-Joint and/or to diagnose SI-Joint pain. Additionally, several non-surgical technologies have been introduced
in the past 10 years to address patients who do not respond to conservative options, including systemic oral medications, opioids, physical
therapy and injection therapy.
To
date, the penetration of a surgical solution for this market has been relatively low (5-7%). We believe this is due to complex surgical
approaches and suboptimal implant design of existing options. The penetration of this market with an optimized surgical solution is Tenon’s
focus.
We
believe the SI-Joint is the last major joint to be successfully addressed by the spine implant industry. Studies have shown that disability
resulting from disease of the SI-Joint is comparable to the disability associated with a number of other serious spine conditions, such
as knee and hip arthritis and degenerative disc disease, each of which has surgical solutions where an implant is used, and a multi-billion-dollar
market exists.
The
SI-Joint
The
SI-Joint is a strong weight bearing synovial joint situated between the lumbar spine and the pelvis and is aligned along the longitudinal
load bearing axis of the human spine when in an upright posture. It functions as a force transfer conduit where it transfers axial loads
bi-directionally from the spine to the pelvis and lower extremities and allows forces to be transmitted from the extremities to the spine.
It also provides load sharing between the hip and spine to contribute towards attenuation of impact shock and stress from activities
of daily living.
The
SI-Joint is a relatively immobile joint that connects the sacrum (the spinal segment that is attached to the base of the lumbar spine
at the L5 vertebra) and the ilium of the pelvis. Each SI-Joint is approximately 2-4mm wide and irregularly shaped.
Motion
of the SI-Joint features vertical shear and rotation. Although the rotational forces about the SI-Joint are relatively low, repetitive
motions created by daily activities such as walking, jogging, twisting at the hips, and jumping can increase the stresses on the SI-Joint.
If the SI-Joint is compromised through injury or degeneration, the load bearing and motion restraints from the surrounding anatomical
structures of the SI-Joint will be compromised resulting in abnormal stress transfers across the joint to these structures, thereby further
augmenting the degenerative cascade of the SI-Joint. Eventual pain and cessation of an individual’s normal activities due to a
painful and unstable SI-Joint have led to an increase in the recent development of SI-Joint stabilization devices.
Non-Surgical
Treatment of Sacroiliac Joint Disease
Several
non-surgical treatments exist for suspected sacroiliac joint pain. These conservative steps often provide desired relief for the patient.
Non-surgical treatments include:
| ● | Drug
Therapy: including opiates and non-steroidal anti-inflammatory medications. |
| ● | Physical
Therapy: which can involve exercises as well as massage. |
| ● | Intra-Articular
Injections of Steroid Medications: which are typically performed by physicians who specialize in pain treatment or anesthesia. |
| ● | Radiofrequency
Ablation: or the cauterizing of the lateral branches of the sacral nerve roots. |
When
conservative steps fail to deliver sustained pain relief and return to quality of life, specific diagnostic protocols are utilized to
explore if a surgical option should be considered.
Diagnosis
Historically,
diagnosing pain from the SI-Joint was not routinely a focus of orthopedic or neurosurgery training during medical school or residency
programs. Due to its invasiveness, post-operative pain, and muscle disruption along with a difficult procedure overall, the open SI-Joint
fusion procedure was rarely taught in these settings.
The
emergence of various SI-Joint surgical technologies has generated a renewed discussion of SI-Joint issues. Of particular focus is the
diagnostic protocol utilized to properly select patients for SI-Joint surgery. Patients with low back pain typically start with primary
care physicians who often refer to pain specialists. Here, the patient will undergo traditional physical therapy combined with oral medications
(anti-inflammatory, narcotic, etc.). If the patient fails to respond to these steps the pain specialist may move to therapeutic injections
of the SI-Joint. These injections may serve to lessen inflammation to the point that the patient is satisfied. However, the impact from
these injections is often transient. In this case the patient is often referred to a clinician to determine if the patient may be a candidate
for surgical intervention. A series of provocative tests in clinic, combined with a specific injection protocol to isolate the SI-Joint
as the pain generator is then utilized to confirm the need for surgical intervention. Published literature has shown this technique to
be a very effective step to determine the best treatment to alleviate pain.
Limitations
of Existing Treatment Options
Surgical
fixation and fusion of the SI-Joint with an open surgical technique was first reported in 1908, with further reports in the 1920s. The
open procedure uses plates and screws, requires a 6 to 12-inch incision and is extremely invasive. Due to the high invasiveness and associated
morbidity, the use of this procedure is limited to cases involving significant trauma, tumor, etc.
Less
invasive surgical options along with implant design began to emerge over the past 15 years. These options feature a variety of approaches
and implant designs and have been met with varying degrees of adoption. Lack of a standard and accepted diagnostic approach, complexity
of approach, high morbidity of approach, abnormally high complication rates and inability to radiographically confirm fusion have all
been cited as reasons for low adoption of these technologies.
The
Market
Based
on market research and internal estimates, we believe the potential market for surgical intervention of the SI-Joint to be 279,000 procedures
annually in the U.S. alone, for a potential annual market of more than $2.2 billion. These estimates are driven by coding data for SI-Joint
injections to treat pain and informed assumptions relative to surgical intervention candidacy.
Based
on public information, we believe that the largest clinical device supplier in this market does approximately 10-11,000 SI-Joint fixations
a year representing the largest market share. The other competitive devices that are offered are all products generally part of much
larger companies with a variety of orthopedic devices and as such do not specifically call out the number of specific SI-Joint procedures
performed with their products. It is our belief that all other competitive devices represent approximately another 5,000 potential SI-Joint
procedures.
Based
on this analysis we believe the market is vastly underserved and only penetrated 5-7%, leaving tremendous upside for a next generation
device that meets the needs of this market.
Competitive
Landscape
We
believe we are the first company to develop and manufacture a novel Inferior Posterior approach featuring a dual pontoon fixation technology
cleared by the FDA expressly for SI-Joint fusion. The approach, referred to as Inferior Posterior Sacroiliac Fusion is focused on these
critical aspects of the surgical procedure:
| 1. | Designed
for Safety: the approach trajectory and angle are away from the neural foramen. |
| 2. | Focus
on Efficiency: the approach is designed to be direct to the SI-Joint, which allows for visualization of the joint and is designed to
pass through minimal muscle structures, which may result in a faster and more efficient surgical procedure and reduced post-op pain for
the patient. |
| 3. | Targeted
Anatomy: the approach places the implant in the aspect of the SI-Joint with the densest bone, designed to provide maximum fixation and
resistance to vertical shear. This is designed to provide a secure press fit of the implant, reducing the incidence of revision surgery
due to implant loosening, which we believe is the reason for many competitive device failures as reported to the FDA Medical Device Reporting
(MDR). |
Note
the trajectory used in the Inferior Posterior approach:
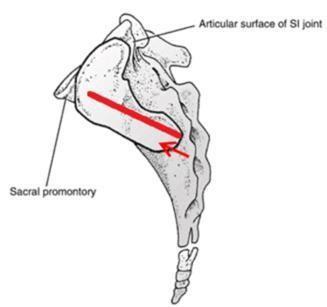
Over
the past several years, other companies have recognized the opportunity and have entered the minimally invasive SI-Joint fixation market.
However, these products are either screw/triangular rod-based or allograft products, which we believe have disadvantages when compared
to The Catamaran System.
In
the United States, we believe that our primary competitors will be SI-Bone, Inc., Globus Medical, Inc., Medtronic plc and RTI Surgical,
Inc. We also compete against non-hardware products, such as allograft bone implants. These allograft products are comprised of human
cells or tissues and are regulated by the FDA differently from implantable medical devices made of metallic or other non-tissue-based
materials. The following chart is a comparison of specifications and features among the various available clinical devices:
Current
Clinical Device Comparison - SI-Joint
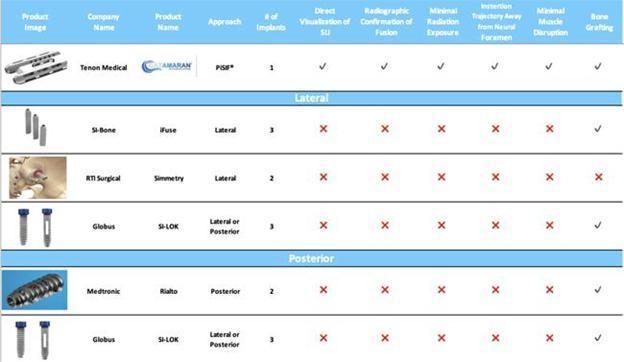
We
believe from our study of the market that many physicians who have been trained to use one of the existing clinical devices have not
adopted the procedure for a variety of reasons. Complexity of approach, high morbidity of approach, abnormally high complication rates
and inability to radiographically confirm fusion have all been cited as reasons for low adoption of these technologies.
The
following are the primary factors on which companies compete in our industry:
| |
● |
product and clinical procedure
effectiveness; |
| |
|
|
| |
● |
ease of surgical technique
and use of associated instruments; |
| |
|
|
| |
● |
safety; |
| |
|
|
| |
● |
published clinical outcomes
and evidence; |
| |
|
|
| |
● |
sales force knowledge and
service levels; |
| |
|
|
| |
● |
product support and service,
and customer service; |
| |
|
|
| |
● |
comprehensive training,
including disease, anatomy, diagnosis, and treatment; |
| |
|
|
| |
● |
product innovation and
the speed of innovation; |
| |
|
|
| |
● |
intellectual property; |
| |
|
|
| |
● |
accountability and responsiveness
to customers’ demands; |
| |
|
|
| |
● |
pricing and reimbursement; |
| |
|
|
| |
● |
scientific (biomechanics)
data; and |
| |
|
|
| |
● |
attracting and retaining
key personnel. |
We
believe that refined approaches and improved implant design will open the door to enhanced adoption and further penetration of this important
market.
The
Catamaran™ SI-Joint Fusion System Solution
Until
October 2022, we sold The Catamaran™ SI-Joint Fusion System to a limited number of clinician advisors to refine the product for
a full commercial launch. In October 2022, we initiated a full commercial launch at the NASS meeting in Chicago. The Catamaran System
includes instruments and implants designed to prepare and fixate the SI-Joint for fusion. We believe The Catamaran System will address
a large market opportunity with a superior product and is distinct from other competitive offerings in the following ways:
| |
● |
Transfixes the SI joint |
| |
● |
Inferior Posterior Sacroiliac
Fusion Approach (PiSIF™) |
| |
● |
Reduced Approach Morbidity |
| |
● |
Direct And Visualized Approach
to the SI-Joint |
| |
● |
Single Implant Technique |
| |
● |
Insertion Trajectory Away
from the Neural Foramen |
| |
● |
Insertion Trajectory Away
from Major Vascular Structures |
| |
● |
Autologous Bone Grafting
in the Ilium, Sacrum and Bridge |
| |
● |
Radiographic Confirmation
of Bridging Bone Fusion of the SI-Joint |
The
fixation device and its key features are shown below:

|
Key
Features
“Pontoon”
in the ilium
“Pontoon”
in the sacrum
“Pontoons
and Bridge” filled with autologous bone from drilling process
Leading
edge osteotome creates defect and facilitates ease of insertion |
The
Catamaran System is a singular implant designed with several proprietary components which allow for it to be explicitly formatted to
transfix the SI-Joint with a single approach and implant. This contrasts with several competitive implant systems that require multiple
approach pathways and implants to achieve fixation. In addition, the Inferior Posterior approach is designed to be direct to the joint
and through limited anatomical structures which may minimize the morbidity of the approach. The implant features a patented dual pontoon
open cell design which enables the clinician to pack the pontoons with the patient’s own autologous bone designed to promote bone
fusion across the joint. The Catamaran System is designed specially to resist vertical shear and rotation of the joint in which it was
implanted, helping stabilize the joint in preparation for eventual fusion.
The
instruments we have developed are proprietary to The Catamaran System and specifically designed to facilitate an Inferior Posterior approach
that is unique to the system.
We
also have developed a proprietary 2D placement protocol as well as a protocol for 3D navigation utilizing the latest techniques in spine
surgery. These Tenon advancements are intended to further enhance the safety of the procedure and encourage more physicians to adopt
the procedure.
The
Catamaran System, as mentioned previously, is placed in the densest aspect of the SI-Joint as confirmed by the pre-op planning images
below:
|
|
|
Surgical
Plan Key:
Yellow:
Guidewire
Purple:
Lateral Pontoon (Ilium)
Green:
Medial Pontoon (Sacrum)
Notes:
Upper
Right Quadrant: The green and purple pontoons represent the placement in the dense bone inferior - contrasted with the dorsal gap
superiorly where competitive systems are most often placed.
Lower
Right Quadrant: The yellow and purple outlines represent The Catamaran System pontoons, illustrating the angle of insertion is away
from the sacral neuro foramen providing for a much safter trajectory for device implantation. |
The
Procedure
We
believe The Catamaran System and its differentiated characteristics allow for an efficient and effective procedure designed to deliver
short-term stabilization and long-term fusion that can be confirmed radiographically. Shown below is an illustration demonstrating the
unique placement of The Catamaran System inserted Inferior Posterior and coming directly down to and transfixing the joint.
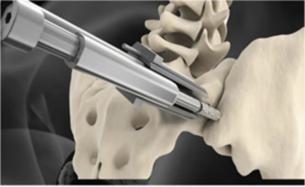 |
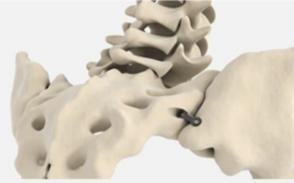 |
The
Catamaran System procedure is typically performed under general anesthesia using a specially designed instrument set we provide to prepare
for the Inferior Posterior access to the SI-Joint. Specially designed imaging and navigation protocols are designed to ensure the clinician
has the proper Entry Point, Trajectory, Angle and Depth (ETAD™) so that the pontoons of The Catamaran System are placed for maximum
fixation. The Catamaran System incorporates two pontoons and is designed so that when the system is impacted into the bone one pontoon
is on the Illum side and the other is in the Sacrum side with the bridge spanning the joint, preventing shear and rotation of the joint.
The device also features an open cell design where the patient’s own (autologous) bone is packed into the pontoons and the bridge
to facilitate fusion across the joint. The leading edge of the bridge is designed to act as an osteotome, providing a self-created deficit
upon insertion. These features are designed to create an ideal environment for bone ingrowth and fusion. Below is a fluoroscopic image
of an implanted Catamaran Fixation Device spanning the SI-Joint.
We
believe the surgical approach and implant design it has developed, along with the 2D and 3D protocols for proper implantation will be
received well by the clinician community who have been looking for a next generation device. Our initial clinical results indicate that
The Catamaran System is promoting fusion across the joint as evidenced by post-op CT scans (the recognized gold standard widely accepted
by the Clinical community).
Post-Op fluoroscopic image
of
implant spanning the SI-Joint |
|
6-Month CT-Scan showing
clear
bridging bone fusion |
| |
|
|
 |
|
 |
A
preliminary 18 case series (Michael Joseph Chaparro, MD, F.A.A.N.S., F.A.C.S.) has documented that The Catamaran System does in fact
promote fusion across the SI-Joint, which many of our competitors have not been able to demonstrate. While products from some of our
competitors use screws and triangular wedges to treat the SI-Joint, most do not effectively resist the vertical shear and twisting within
the joint. This 18 patient series was presented at the North American Spine Society Annual Meeting in Chicago, IL in October 2022.
An
independent biomechanical study (Lisa Ferrara, Ph.D. OrthoKinetic Technologies, LLC now part of Element) demonstrated that a single Catamaran
SIJ Fixation Device was superior to predicate device in the areas of Fixation Strength, Shear Stiffness, Dynamic Endurance and Pullout
Strength. We hold issued patents on The Catamaran System and its unique features including the dual pontoons and the open cell structure
for bone graft packing. We also hold an issued patent for the method of placing The Catamaran System into the SI-Joint where one pontoon
is in the ilium and the other in the sacrum.
The
Catamaran System’s unique design has already demonstrated radiographically confirmed fusion in initial patients. We believe that
this beneficial advantage along with a simpler, safer, and less painful procedure will make this the procedure of choice for most physicians.
We have initiated post market, IRB controlled clinical trials to demonstrate this technology delivers on these advantages.
Coverage
and Reimbursement
When
a Tenon procedure utilizing The Catamaran System is performed, the healthcare facility, either a hospital (inpatient or outpatient clinic),
and the clinician submit claims for reimbursement to the patient’s insurer. Generally, the facility obtains a lump sum payment,
or facility fee, for SI-Joint fusions. Our products are purchased by the facility, along with other supplies used in the procedure. The
facility must also pay for its own fixed costs of operation, including certain operating room personnel involved in the procedure, ICD
and other medical services care. If these costs exceed the facility reimbursement, the facility’s managers may discourage or restrict
clinicians from performing the procedure in the facility or using certain technologies, such as The Catamaran System, to perform the
procedure.
The
Medicare 2023 national average hospital inpatient payment for SI-Joint procedures ranges from approximately $25,661 to approximately
$46,437 depending on the procedural approach and the presence of Complication and Comorbidity/Major Complication and Comorbidity.
The
Medicare 2023 national average hospital outpatient clinic payment is $17,756. We believe that insurer payments to facilities are generally
adequate for these facilities to offer The Catamaran System procedure.
Physicians
are reimbursed separately for their professional time and effort to perform a surgical procedure. Depending on the surgical approach,
the incision size, type and extent of imaging guidance, indication for procedure, and the insurer, The Catamaran System procedure may
be reported by the physician using any one of the applicable following CPT® codes 27279, 27280, 27299. The Medicare 2022 national
average payment for CPT® 27279 is $807 and $1,352 for 27280. CPT® 27299 has no national valuation. Clinicians, however, can present
a crosswalk to another procedure believed to be fairly equivalent and/or comparison to a code for which there is an existing valuation.
For
some governmental programs, such as Medicaid, coverage and reimbursement differ from state to state, and some state Medicaid programs
may not pay an adequate amount for the procedures performed with our products, if any payment is made at all. Similar to Medicaid, many
private payors’ coverage and payment may differ from one payer to another.
We
believe that some clinicians view the current Medicare reimbursement amount as insufficient for current SI-Joint procedures, given the
work effort involved with the procedure, including the time to diagnose the patient and obtain prior authorization from the patient’s
health insurer when necessary. Many private payors require extensive documentation of a multi-step diagnosis before authorizing SI-Joint
fusion for a patient. We believe that some private payors apply their own coverage policies and criteria inconsistently, and clinicians
may experience difficulties in securing approval and coverage for sacroiliac fusion procedures. Additionally, many private payors limit
coverage for open SI-Joint fusion to trauma, tumors or extensive spine fusion procedures involving multiple levels.
We
believe the unique design of The Catamaran System and the fact The Catamaran System may be placed both via an open procedure based on
the clinician’s determination of trauma induced SI-Joint pain or as a minimally invasive approach provides a unique and differentiated
approach for the clinician to determine the reimbursement code that best fits the clinical problem. We believe this is a significant
advantage over competitive devices by providing the clinician the clinical flexibility of offering the best clinical solution and approach
for patients.
Sales
and Marketing
We
will market and sell The Catamaran System primarily through independent distributors and sales representatives specializing in orthopedics
and spine sales. Our target customer base includes approximately 12,000 physicians who perform spine and/or pelvic surgical procedures.
We
will provide general sales and marketing training to our independent sales representative along with comprehensive, hands-on cadaveric
and dry-lab training sessions focusing on the clinical benefits of The Catamaran System and the importance of using the 2D and 3D protocols
we have developed. We believe many clinicians have already been trained using one of the alternative products but have not been satisfied
with the approach and technology. This provides us with an opportunity to demonstrate to an already-trained-clinician the unique attributes
of The Catamaran System.
Our
business objective is to introduce the Next Generation Implant for SI-Joint Fixation. The past 10 years has seen an acceleration in recognition
and discussion of the SI-Joint as a cause of pain that can be treated. However, adoption has been hindered by complexity of the procedure
as evidenced by the significant number of reported Medical Device Records (MDR’s). The need for multiple implants and resulting
post-op pain has also contributed to low adoption numbers. Our strategy is to provide a safer, faster, and better surgical experience
and a significant pain reduction benefit for the patient. Our goals are simple but impactful and as such we plan on the following:
| |
● |
Educate and inform physicians
and other healthcare providers, payors, and patients about the growing body of evidence supporting what we believe is the safety,
durable clinical effectiveness, economic benefit, and reduction in opioid use associated with SI-Joint fixation and The Catamaran
System procedure. |
| |
|
|
| |
● |
Utilize the most effective
means of training via video and in-person labs demonstrating the ease of use with 2D and 3D navigation. Since many physicians have
already been trained but have not incorporated SI-Joint fixation into their practices we will work with these physicians to reengage
and train them on the Next Generation of an SI-Joint implant which incorporates a safer and simpler approach. |
| |
|
|
| |
● |
Utilize the best approaches
of direct-to-consumer outreach to educate patients that there is a safe solution to help them improve their quality of life. Additionally,
to reach the broadest physician and patient audience on case study results from around the United States we plan to implement an
active social media campaign incorporating Facebook, Instagram, YouTube, etc. |
| |
|
|
| |
● |
Invest in our independent
sales representative network to ensure that all Tenon representatives have the latest in marketing and education tools to reduce
the time from training to adoption. |
| |
|
|
| |
● |
Remain true to our next
generation product development strategy by continually bringing out new advancements in and around the SI-Joint and pelvic region. |
| |
|
|
| |
● |
Continue to grow our existing intellectual property
portfolio. |
| |
|
|
| |
● |
Execute post-market clinical research to confirm the
benefits of the distinct approach and implant. |
Regulatory
Status
We
have received FDA 510(k) clearance to market and sell The Catamaran System for sacroiliac joint fusion for conditions including sacroiliac
joint disruptions and degenerative sacroiliitis.
Research
& Development
Our
initial development of The Catamaran System has incorporated several differentiating features which we believe will make an important
contribution for many patients suffering from SI-Joint pain. To our knowledge no other competitive product incorporates these Next Generation
features:
| |
● |
Dual Pontoon implant that transfixes the targeted joint; |
| |
|
|
| |
● |
Open cell design designed for utilizing the patient’s
own autologous bone for promotion of fusion; |
| |
|
|
| |
● |
Bridge design between the dual pontoons for enhanced
strength; |
| |
|
|
| |
● |
Leading edge of the implant
designed to function as an osteotome providing a self-creating defect feature not available with competitive systems; |
| |
|
|
| |
● |
Single implant designed with varying pontoon sizes
to ensure a robust fixation based on anatomy; and |
| |
|
|
| |
● |
Additional smaller Catamaran designed for smaller anatomy
and/or revision surgery. |
The
Tenon development plan is to expand The Catamaran System offering by introducing a series of progressively longer pontoons so that the
clinician has a full complement of sized implants to choose from depending on the patient’s anatomy. These product enhancements
will enable the clinician to optimize the size of each implant to ensure full fixation based on anatomy. We believe, based on literature
searches of prior SI-Joint fixation technologies, that adverse event incidence where the implant has loosened or been misplaced thereby
requiring a revision surgery could reach 20%. We believe that our ability to make The Catamaran System a specifically sized fixation
device will benefit many patients requiring a revision surgery.
The
Catamaran System shown below has been cleared by the FDA for commercialization. This patented titanium implant incorporates The Catamaran
SI-Joint Fixation Device pontoon design and the open cell configuration which we believe, when filled with the patient’s autologous
bone, promotes fusion. The two images below show a comparison of a competitive implant requiring three implants and The Catamaran System
unique pontoon design showing the need of only one implant to cover the same amount of the SI-Joint.
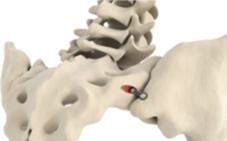 |
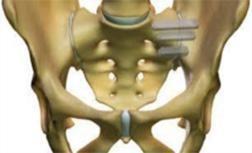 |
| |
|
The Catamaran™ SIJ
Fusion
System Single Implant |
SI
Bone iFuse® Three Implants
|
Our
mission will be to continue developing enhancements to The Catamaran System to meet our customers’ changing needs and to improve
the surgery’s effectiveness. This includes revision surgery options as well as options as an adjunct to long fusion constructs
in the lumbar spine.
Additionally,
we will initiate various post marketing clinical studies in accordance with FDA cleared indications for use. Since we have already received
FDA 510(k) clearance to market The Catamaran System, our clinical study activities will be focused on capturing post-market safety and
efficacy data. Tenon has received IRB approval for two post-market trials, including a 50 patient, 10 center multi-center trial and a
prospective CT trial to demonstrate fusion in patient who have already been treated with The Catamaran System. Clinical study endpoints
may include but are not limited to; pain scoring, length of surgical procedure, blood loss, post-op pain, length of stay, duration of
non-weight-bearing post-op, radiographic confirmation of fusion and surgical complication rates. Statistical analysis plans may be designed
to demonstrate non-inferiority to historical control, as reported in published literature, which may be used for submission to peer reviewed
articles/posters/presentations and the like.
Intellectual
Property
Developing
and maintaining a strong intellectual property position is an important element of our business. We maintain the intellectual property
through a combination of patent protection, trademarks, and trade secrets. We have sought, and will continue to seek, patent protection
for our technology, for improvements to our technology, as well as for any of our other technologies where we believe such protection
will be advantageous.
As
of August 13, 2024, we own four (4) issued U.S. utility patents, eighteen (18) pending U.S. utility patent applications, four (4) issued
foreign utility patents in Australia, Canada, Japan and Israel, and two (2) pending foreign utility patent applications in the European
Community, Brazil and Japan. We also have thirteen (13) registered trademarks (seven (7) U.S. and six (6) foreign) and twelve (12) pending
trademark applications in the U.S.
Our
utility patents and patent applications are directed to several different aspects of our sacroiliac (SI) joint stabilization technology
and related patent platform. By way of example, our granted patents and pending patent applications cover various structural features
of our unique Catamaran SI-Joint prosthesis and means for employing same to stabilize a dysfunctional SI-Joint.
The
term of individual patents depends on the legal term for patents in the countries in which they are granted. In most countries, including
the United States, the patent term for a utility patent is generally 20 years from the earliest claimed filing date of a nonprovisional
patent application in the applicable country. Our issued U.S. and foreign utility patents are anticipated to naturally expire around
2031, and our U.S. pending utility patent applications, if issued into patents, are similarly anticipated to naturally expire around
2031, excluding any additional patent term adjustment(s) or extension(s), and assuming payment of all applicable maintenance or annuity
fees. Once a patent expires, patent protection ends and an invention enters the public domain allowing anyone to commercially exploit
the invention without infringing the patent.
We
cannot guarantee that patents will be issued from any of our pending applications or that issued patents will be of sufficient scope
or strength to provide meaningful protection for our technology. Notwithstanding the scope of the patent protection available to us,
a competitor could develop methods or devices that are not covered by our patents or circumvent these patents. Furthermore, although,
at present, we are unaware of any patent applications that may result in one or more issued patents that our existing products or technologies
may be alleged to infringe, since U.S. and foreign applications can take many months to publish, there may be applications unknown to
us that may result in one or more issued patents that our existing products or technologies may be alleged to infringe.
As
of August 13, 2024, we also have priority rights in and to several significant trademarks that support our products and brand, including
seven (7) registered U.S. trademarks, twelve (12) U.S. trademark applications and six (6) foreign trademark applications in the European
Community (excluding the United Kingdom), Australia and Japan.
Regulation
Domestic Regulation of Our Products and
Business. Our research, development and clinical programs, as well as our manufacturing and marketing operations, are subject
to extensive regulation in the United States and other countries. Most notably, all of our products sold in the United States are subject
to the federal Food, Drug and Cosmetic Act (the “FDCA”), as implemented and enforced by the FDA. The FDA governs the following
activities that we perform or that are performed on our behalf, to ensure that medical products distributed domestically or exported internationally
are safe and effective for their intended uses:
| ● | product
design, development, and manufacture; |
| ● | product
safety, testing, labeling, and storage; |
| ● | record
keeping procedures; |
| ● | product
marketing, sales, distribution and export; and |
| ● | post-marketing
surveillance, complaint handling, medical device reporting, reporting of deaths, serious injuries or device malfunctions, and repair
or recall of products. |
There are numerous FDA regulatory requirements
governing the clearance or approval and marketing of our products. These include:
| ● | product
listing and establishment registration, which helps facilitate FDA inspections and other regulatory action; |
| ● | investigational
device exemptions to conduct premarket clinical trials, which include extensive monitoring, recordkeeping, and reporting requirements; |
| ● | QSR,
which requires manufacturers, including contract manufacturers, to follow stringent design, testing, control, documentation and other
quality assurance procedures during all aspects of the manufacturing process; |
| ● | labeling
regulations and FDA prohibitions against the promotion of products for uncleared, unapproved or off-label use or indication; |
| ● | clearance
of product modifications that could significantly affect safety or effectiveness or that would constitute a major change in intended
use of one of our cleared devices; |
| ● | approval
of product modifications that affect the safety or effectiveness of one of our approved devices; |
| ● | medical
device reporting regulations, which require that manufacturers comply with FDA requirements to report if their device may have caused
or contributed to a death or serious injury, or has malfunctioned in a way that would likely cause or contribute to a death or serious
injury if the malfunction of the device or a similar device were to recur; |
| ● | post-approval
restrictions or conditions, including post-approval study commitments; |
| ● | post-market
surveillance regulations, which apply when necessary to protect the public health or to provide additional safety and effectiveness data
for the device; |
| ● | the
FDA’s recall authority, whereby it can ask, or under certain conditions order, device manufacturers to recall from the market a
product that is in violation of governing laws and regulations; |
| ● | regulations
pertaining to voluntary recalls; and |
| ● | notices
of corrections or removals. |
The FDA has broad post-market and regulatory enforcement
powers. We and our contract manufacturers are subject to announced and unannounced inspections by the FDA to determine our compliance
with the QSR and other regulations and these inspections may include the manufacturing facilities of our suppliers. Tenon has a robust
Supplier Qualification and Audit process as part of our quality system that ensures contract manufacturers, and their suppliers meet all
requirements.
An FDA pre-approval inspection is not required
for The Catamaran System due to its lower device classification, class II versus the higher class III. As is the case for most medical
device firms, Tenon is subject to routine and “for cause” FDA inspections. Routine inspections are mandated by
law every 2 years for class II and class III device manufacturers and make up the majority of FDA’s inspections. If a serious public
health risk is identified during a routine inspection, the inspection may convert to a “for cause” inspection. In the current
environment, FDA has limited compliance resources and has not been able to perform routine inspections in accordance with the 2-year mandate.
Therefore, FDA uses a risk-based approach when deciding which firms should be selected for a routine inspection. Using the Establishment
Registration and Device Listing databases, FDA identifies who manufactures and/or distributes which devices. The firms are then prioritized
by risk, class III > class II > class I. Firms that have recently introduced a new device to the market also are given higher priority,
as well as those that have had significant prior violations and complaints. At present, Tenon has not been selected for an FDA inspection.
Tenon uses best practices to secure and maintain regulatory compliance by engaging with suppliers and contract manufacturing firms that
are ISO 13485 (or equivalent) compliant and by periodically performing internal, external, and third-party inspections and audits of the
facilities and systems to assess compliance.
FDA Premarket Clearance and Approval Requirements.
Unless an exemption applies, each medical device we wish to commercially distribute in the United States will require either premarket
notification, or 510(k), clearance or approval of a PMA from the FDA. The FDA classifies medical devices into one of three classes. Devices
deemed to pose lower risks are placed in either Class I or II, which typically requires the manufacturer to submit to the FDA a premarket
notification requesting permission to commercially distribute the device. This process is generally known as 510(k) clearance. Some low-risk
devices are exempted from this requirement. Devices deemed by the FDA to pose the greatest risks, such as life-sustaining, life- supporting
or implantable devices, or devices deemed not substantially equivalent to a previously cleared 510(k) device, are placed in Class III,
requiring a PMA. If the FDA agrees that the device is substantially equivalent to a predicate device currently on the market, it will
grant 510(k) clearance to commercially market the device. If the FDA determines that the device is “not substantially equivalent”
to a previously cleared device, the device is automatically designated as a Class III device. The device sponsor must then fulfill more
rigorous PMA requirements or can request a risk-based classification determination for the device in accordance with the “de novo”
process, which is a route to market for novel medical devices that are low to moderate risk and are not substantially equivalent to a
predicate device. All of our currently marketed products are Class II devices, subject to 510(k) clearance.
After a device receives 510(k) marketing clearance,
any modification that could significantly affect its safety or effectiveness, or that would constitute a major change or modification
in its intended use, will require a new 510(k) marketing clearance or, depending on the modification, PMA approval. The determination
as to whether or not a modification could significantly affect the device’s safety or effectiveness is initially left to the manufacturer
using available FDA guidance. Many minor modifications today are accomplished by a “letter to file” in which the manufacture
documents the rationale for the change and why a new 510(k) is not required. However, the FDA may review such letters to file to evaluate
the regulatory status of the modified product at any time and may require the manufacturer to cease marketing and recall the modified
device until 510(k) clearance or PMA approval is obtained. The manufacturer may also be subject to significant regulatory fines or penalties.
Clinical Trials. Clinical trials
are generally required to support a PMA application and are sometimes required for 510(k) clearance. Such trials for implanted devices
such as The Catamaran SIJ Fixation Device generally require an investigational device exemption application, or IDE, approved in advance
by the FDA for a specified number of subjects and study sites, unless the product is deemed a nonsignificant risk device eligible for
more abbreviated IDE requirements. Clinical trials are subject to extensive monitoring, recordkeeping, and reporting requirements. Clinical
trials must be conducted under the oversight of an institutional review board, or IRB, for the relevant clinical trial sites and must
comply with FDA regulations, including but not limited to those relating to good clinical practices. To conduct a clinical trial, we also
are required to obtain the subjects’ informed consent in form and substance that complies with both FDA requirements and state and
federal privacy and human subject protection regulations. We, the FDA, or the institutional review board, or IRB, could suspend a clinical
trial at any time for various reasons, including a belief that the risks to study subjects outweigh the anticipated benefits. Even if
a trial is completed, the results of clinical testing may not adequately demonstrate the safety and effectiveness of the device or may
otherwise not be sufficient to obtain FDA clearance or approval to market the product in the United States.
Pervasive and Continuing Regulation. After
a device is placed on the market, numerous regulatory requirements continue to apply. These include:
| ● | Product
listing and establishment registration, which helps facilitate FDA inspections and other regulatory action; |
| ● | QSR,
which requires manufacturers, including contract manufacturers, to follow stringent design, testing, control, documentation, and other
quality assurance procedures during all aspects of the manufacturing process; |
| ● | labeling
regulations and FDA prohibitions against the promotion of products for uncleared, unapproved, or off-label use or indication; |
| ● | clearance
of product modifications that could significantly affect safety or effectiveness or that would constitute a major change in intended
use of one of our cleared devices; |
| ● | approval
of product modifications that affect the safety or effectiveness of one of our approved devices; |
| ● | post-approval
restrictions or condition, including post-approval study commitments; |
| ● | post-market
surveillance regulations, which apply when necessary to protect the public health or to provide additional safety and effectiveness data
for the device; |
| ● | the
FDA’s recall authority, whereby it can ask, or under certain conditions order, device manufacturers to recall from the market a
product that is in violation of governing laws and regulations; |
| ● | regulations
pertaining to voluntary recalls; and |
| ● | notices
of corrections or removals. |
The FDA has broad post-market and regulatory enforcement
powers. We are subject to unannounced inspections by the FDA to determine our compliance with the QSR and other regulations, and these
inspections may include the manufacturing facilities of some of our subcontractors. Failure by us or by our suppliers to comply with applicable
regulatory requirements can result in enforcement action by the FDA or other regulatory authorities, which may result in sanctions including,
but not limited to:
| ● | untitled
letters, warning letters, fines, injunctions, consent decrees, and civil penalties; |
| ● | unanticipated
expenditures to address or defend such actions |
| ● | customer
notifications for repair, replacement, refunds; |
| ● | Recall,
detention, or seizure of our products; |
| ● | operating
restrictions or partial suspension or total shutdown of production; |
| ● | refusing
or delaying our requests for 510(k) clearance or PMA approval of new products or modified products; |
| ● | withdrawing
510(k) clearances or PMA approvals that have already been granted: |
| ● | refusal
to grant export approval for our products; or |
The FDA has not yet inspected our contract manufacturer’s manufacturing
facilities.
Promotional Materials “Off-Label”
Promotion. Advertising and promotion of medical devices, in addition to being regulated by the FDA, are also regulated by the
Federal Trade Commission and by state regulatory and enforcement authorities. If the FDA determines that our promotional materials or
training constitutes promotion of an unapproved use, it could request that we modify our training or promotional materials or subject
us to regulatory or enforcement actions, including the issuance of an untitled letter, a warning letter, injunction, seizure, civil fine,
or criminal penalties. It is also possible that other federal, state, or foreign enforcement authorities might take action if they consider
our promotional or training materials to constitute promotion of an unapproved use, which could result in significant fines or penalties
under other statutory authorities, such as laws prohibiting false claims for reimbursement. In that event, our reputation could be damaged,
and adoption of the products would be impaired.
In addition, under the federal Lanham Act and
similar state laws, competitors, and others can initiate litigation relating to advertising claims.
Healthcare Fraud and Abuse
Federal and state governmental agencies and equivalent
foreign authorities subject the healthcare industry to intense regulatory scrutiny, including heightened civil and criminal enforcement
efforts. These laws constrain the sales, marketing and other promotional activities of medical device manufacturers by limiting the kinds
of financial arrangements we may have with hospitals, physicians and other potential purchases of our products. Federal healthcare fraud
and abuse laws apply to our business when a customer submits a claim for an item or service that is reimbursed under Medicare, Medicaid,
or other federally funded healthcare programs. Descriptions of some of the laws and regulations that may affect our ability to operate
follows.
The federal Anti-Kickback Statute prohibits, among
other things, persons from knowingly and willfully soliciting, receiving, offering, or paying remuneration, directly or indirectly, in
cash or in kind, to induce or reward either the referral of an individual for, or the purchase, order or recommendation of, items or services
for which payment may be made, in whole or in part, under federal healthcare programs. The term “remuneration” has been broadly
interpreted to include anything of value, and the government can establish a violation of the Anti-Kickback Statute without proving that
a person or entity had actual knowledge of, or a specific intent to violate, the law. The Anti-Kickback Statute is subject to evolving
interpretations and has been applied by government enforcement officials to a number of common business arrangements in the medical device
industry. There are a number of statutory exceptions and regulatory safe harbors protecting some common activities from prosecution; however,
those exceptions and safe harbors are drawn narrowly, and there is no exception or safe harbor for many common business activities. Failure
to meet all of the requirements of a particular statutory exception or regulatory safe harbor does not make the conduct per se illegal
under the Anti-Kickback Statute, but the legality of the arrangement will be evaluated on a case-by-case basis based on the totality of
the facts and circumstances. A number of states also have anti-kickback laws that establish similar prohibitions that may apply to items
or services reimbursed by government programs, as well as by any third-party payors, including commercial payors.
The civil False Claims Act prohibits, among other
things, knowingly presenting or causing the presentation of a false or fraudulent claim for payment of federal funds, or knowingly making,
or causing to be made, a false record or statement material to a false or fraudulent claim to avoid, decrease or conceal an obligation
to pay money to the federal government. A claim including items or services resulting from a violation of the Anti- Kickback Statute constitutes
a false or fraudulent claim for purposes of the False Claims Act. Actions under the False Claims Act may be brought by the government
or as a qui tam action by a private individual in the name of the government. Qui tam actions are filed under seal and impose
a mandatory duty on the U.S. Department of Justice to investigate such allegations. Most private citizen actions are declined by the Department
of Justice or dismissed by federal courts. However, the investigation costs for a company can be significant and material even if the
allegations are without merit. There are also criminal penalties, including imprisonment and criminal fines, for making or presenting
a false or fictitious or fraudulent claim to the federal government.
False Claims Act liability is potentially significant
in the healthcare industry because the statute provides for treble damages and mandatory penalties of $11,181 to $22,363 per claim (adjusted
annually for inflation). Because of the potential for large monetary exposure, healthcare companies often resolve allegations without
admissions of liability for significant and sometimes material amounts to avoid the uncertainty of treble damages and per claim penalties
that may awarded in litigation proceedings. Moreover, to avoid the risk of exclusion from federal healthcare programs as a result of a
False Claims Act settlement, companies may enter into corporate integrity agreements with the government, which may impose substantial
costs on companies to ensure compliance.
In addition, HIPAA created federal criminal statutes
that prohibit, among other actions, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit
program, including private third-party payors, knowingly and willfully embezzling or stealing from a healthcare benefit program, willfully
obstructing a criminal investigation of a healthcare offense, and knowingly and willfully falsifying, concealing or covering up a material
fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits,
items or services.
The federal Physician Payment Sunshine Act, implemented
by CMS as the Open Payments program, requires manufacturers of drugs, devices, biologics and medical supplies for which payment is available
under Medicare, Medicaid, or the Children’s Health Insurance Program to report annually to CMS information related to payments or
other “transfers of value” made to physicians and teaching hospitals, and requires applicable manufacturers to report annually
to CMS ownership and investment interests held by physicians and their immediate family members and payments or other “transfers
of value” to such physician owners.
Certain states also mandate implementation of
corporate compliance programs, impose restrictions on device manufacturer marketing practices, and/or require tracking and reporting of
gifts, compensation, and other remuneration to healthcare professionals and entities.
The Foreign Corrupt Practices Act and similar
anti-bribery laws in other countries, such as the UK Bribery Act, generally prohibit companies and their intermediaries from making improper
payments to government officials and/or other persons for the purpose of obtaining or retaining business. Our policies mandate compliance
with these anti-bribery laws.
Violations of these federal and state fraud abuse
laws can subject us to administrative, civil, and criminal penalties, including imprisonment, substantial fines, penalties, damages, and
exclusion from participation in federal healthcare programs, including Medicare and Medicaid.
Data Privacy and Security Laws
HIPAA requires the notification of patients, and
other compliance actions, in the event of a breach of unsecured PHI. If notification to patients of a breach is required, such notification
must be provided without unreasonable delay and in no event later than 60 calendar days after discovery of the breach. In addition, if
the PHI of 500 or more individuals is improperly used or disclosed, we could be required to report the improper use or disclosure to the
U.S. Department of Health and Human Services, or HHS, which would post the violation on its website, and to the media. Failure to comply
with the HIPAA privacy and security standards can result in civil monetary penalties up to $55,910 per violation, not to exceed $1.68
million per calendar year for non-compliance of an identical provision, and, in certain circumstances, criminal penalties with fines up
to $250,000 per violation and/or imprisonment.
In addition, even when HIPAA does not apply, according
to the FTC, failing to take appropriate steps to keep consumers’ personal information secure constitutes unfair acts or practices
in or affecting commerce in violation of Section 5(a) of the FTCA, 15 U.S.C § 45(a). The FTC expects a company’s data security
measures to be reasonable and appropriate in light of the sensitivity and volume of consumer information it holds, the size and complexity
of its business, and the cost of available tools to improve security and reduce vulnerabilities. Medical data is considered sensitive
data that merits stronger safeguards. The FTC’s guidance for appropriately securing consumers’ personal information is similar
to what is required by the HIPAA Security Rule.
We are subject to the supervision of local data
protection authorities in those jurisdictions where we are established or otherwise subject to applicable law. We depend on a number of
third parties in relation to our provision of our services, a number of which process personal data on our behalf. With each such provider
we enter into contractual arrangements to ensure that they only process personal data according to our instructions, and that they have
sufficient technical and organizational security measures in place. Where we transfer personal data outside the EEA, we do so in compliance
with the relevant data export requirements. We take our data protection obligations seriously, as any improper disclosure, particularly
with regard to our customers’ sensitive personal data, could negatively impact our business and/or our reputation.
Manufacturing and Supply
We do not manufacture any products or component
parts and currently use five contract manufacturers to produce all of our instruments, implants and sterilization cases. The majority
of our instruments have a secondary manufacturing supplier, and we continually work with additional manufacturers to establish secondary
manufacturing suppliers. Our contract manufacturers source and purchase all raw materials used in the manufacture of The Catamaran System
which includes mainly stainless steel and aluminum for our instruments and sterilization cases and titanium for our implants.
We do not currently have manufacturing agreements
with any of our contract manufacturers and orders are controlled through purchase orders. The Company does not believe its relationship
with any one contract manufacturer is material to its business.
We believe the manufacturing operations of our
contract manufacturers, and those of the suppliers of our manufacturers, comply with regulations mandated by the FDA, as well as Medical
Devices Directive regulations in the EEA. Manufacturing facilities that produce medical devices or component parts intended for distribution
world-wide are subject to regulation and periodic planned and unannounced inspection by the FDA and other domestic and international regulatory
agencies.
In the United States, the product we sell is required
to be manufactured in compliance with the QSR, which covers the methods used in, and the facilities used for, the design, testing, control,
manufacturing, labelling, quality assurance, packaging, storage, and shipping.
We are required to demonstrate continuing compliance
with applicable regulatory requirements and will be subject to FDA inspections. Further, we and certain of our contract manufacturers
are required to comply with all applicable regulations and current good manufacturing practices. As set forth above, these FDA regulations
cover, among other things, the methods and documentation of the design, testing, production, control, quality assurance, labeling, packaging,
sterilization, storage, and shipping of our products. Compliance with applicable regulatory requirements is subject to continual review
and is monitored rigorously through periodic inspections. If we or our manufacturers fail to adhere to current good manufacturing practice
requirements, this could delay production of our products and lead to fines, difficulties in obtaining regulatory approvals, recalls,
enforcement actions, including injunctive relief or consent decrees, or other consequences, which could, in turn, have a material adverse
effect on our financial condition or results of operations.
Product Liability and Insurance
The manufacture and sale of our products subjects
us to the risk of financial exposure to product liability claims. Our products are used in situations in which there is a risk of serious
injury or death. We carry insurance policies which we believe to be customary for similar companies in our industry. We cannot assure
you that these policies will be sufficient to cover all or substantially all losses that we experience.
We endeavor to maintain executive and organization
liability insurance in a form and with aggregate coverage limits that we believe are adequate for our business purposes.
Legal Proceedings
We may also from time to time be, party to litigation
and subject to claims incident to the ordinary course of business. As our growth continues, we may become party to an increasing number
of litigation matters and claims. The outcome of litigation and claims cannot be predicted with certainty, and the resolution of these
matters could materially affect our future results of operations, cash flow or financial position.
Employees
As of August 13, 2024, we had a total of 23 employees,
all of whom are full-time, and three senior consulting advisors of various specialty including product development, clinical affairs reimbursement.
None of our employees is subject to a collective bargaining agreement, and we consider our relationship with our employees to be good.
Property
We lease and maintain our primary offices at 104
Cooper Court, Los Gatos, CA 95032. We do not currently own any real estate.
Corporate Information
We were incorporated on June 6, 2012, in Delaware.
Our principal executive offices are located at 104 Cooper Court, Los Gatos, CA 95032 and our telephone number is (408) 649-5760. Our website
address is www.tenonmed.com. The information on, or that can be accessed through, our website is not part of this prospectus. We
have included our website address as an inactive textual reference only.
MANAGEMENT
The following are our executive officers, principal
accounting officer and directors and their respective ages and positions as of August 13, 2024.
| Name | |
Age | | |
Position |
| Steven M. Foster | |
| 56 | | |
Chief Executive Officer and President, Director |
| Richard Ginn | |
| 58 | | |
Chief Technology Officer and Director |
| Jay Hanson | |
| 58 | | |
Director of SEC Reporting and Compliance (principal accounting officer) |
| Richard Ferrari | |
| 70 | | |
Executive Chairman of the Board |
| Ivan Howard | |
| 57 | | |
Director |
| Kristine M. Jacques | |
| 57 | | |
Director |
| Robert K. Weigle | |
| 64 | | |
Director |
| Stephen H. Hochschuler, M.D. | |
| 81 | | |
Director |
Steven M. Foster is our Chief Executive
Officer and President, and is also a director of the Company. Mr Foster has over 30 years of marketing, sales, operations and general
management experience. From 2015 to present Mr. Foster has been a principal with CTB Advisors, LLC in Brentwood, Tennessee. CTB Advisors
was founded as a single member limited liability company for the purpose of providing medical device organizations and physicians with
consultative assistance on commercialization focused projects. Projects included: CRM based clinician engagement program design, training
and implementation for NuVasive (NUVA). Valuation assessment / business plan development of early-stage spine technology including IP
assessment and regulatory pathway definition. M&A (SafeOp Surgical) integration project, Alphatec Spine (ATEC). Current Status: Exclusive
to ATEC. From 2012 to 2014 Mr. Foster was Global Commercialization President of Safe Orthopedics SAS, Paris, FR (based in Michigan): There
Mr Foster worked on early-stage commercialization of a novel single-use / sterile / traceable surgical kit for lumbar spine fusion. His
focus included pre-clinical design, clinician advisor team development, early marketing, web design, convention presence and P&L preparation
and management. Technology reached 200 global surgeries in first 12 months of commercialization. From 1992 to 2012 Mr. Foster was part
of the Danek Group Inc., Sofamor Danek, Medtronic Spine organization where he held a variety of marketing, sales administration and general
management roles, including as VP / GM of Medtronic Spine’s Western Europe operations from 2007-2010. Mr. Foster received a Bachelor
of Science, Business Administration with a concentration in Marketing and Management from Central Michigan University in 1990.
Richard Ginn is a founder, the Chief Technology
Officer and a director of the Company. Mr. Ginn’s focus is primarily on intellectual property and product development, he has travelled
throughout the world to train physicians and participated in multiple FIH trials and is a named inventor on more than 300 patents for
medical devices. Over the course of his career, he has helped raise more than $100 million in venture capital and has provided an average
10x return to his investors. Mr. Ginn is the founder of TransAortic Medical, an embolic protection device company, and is its President,
CEO and a director from 2013 to present. At TransAortic, Mr. Ginn Managed all corporate operations, raised capital to support company
needs; managed acquisition of technology by strategic partner; managed all Intellectual Property; and set up European distribution for
CE Marked device. Mr. Ginn is the founder of Promed, a large hole femoral closure device company and was the CEO, President and a director
from 2012 to 2019. At Promed he managed all corporate operations; raised capital to support company needs; and managed all intellectual
property.
Jay Hanson is our Director of SEC Reporting
and Compliance. Mr. Hanson has been the Director of SEC Reporting and Compliance since May 22, 2022. From 2020 to 2022 Mr. Hanson was
the Associate Director, SEC Reporting and SOX Compliance at Zosano Pharma, a pharmaceutical company in Fremont, California. From 2014
to 2020, Mr. Hanson was the Associate Director of Accounting and Financial Reporting at Vivus, Inc., a pharmaceutical company in Mountain
View, California. From 2013 to 2014, Mr. Hanson was the Controller at Alexza Pharmaceuticals, Inc., a pharmaceutical company in Mountain
View, California. Mr. Hanson received a Masters in Accountancy with an emphasis in Accounting Information Systems concurrently with a
Bachelor of Science, Accounting from Brigham Young University in 1991.
Richard Ferrari is a founder, a director
and Executive Chairman of the Company. Since 2000, Mr. Ferrari has been and currently is a Managing Director of Denovo Ventures a $650
million venture firm specializing in Medical Devices and Biotechnology. From January 2019 until April 2021 Mr. Ferrari was employed as
CEO and Chairman of the Board of Directors of PQ Bypass which culminated is a successful acquisition by Endologix. During the last five
years Mr. Ferrari has been and currently is a board member (Executive Chairman) of Medlumics, S.L., a medical device company founded in
2011; a board member (Vice Chairman) of ABS Interventional; a board member (Executive Chairman) of Heart Beam Inc.; a board member of
Biomodex Corporation; a board member of Retriever Medical Inc.; a board member of RMx Medical; a board member of Hawthorne Effect, Inc.;
a board member and co-founder of TransAortic acquired by Medtronic; Executive Chairman of Sentreheart acquired by Atricure, a board member
of Spinal Modualtion sold to St Jude and a board member of Hands of Hope. Mr. Ferrari has raised over $1 billion for the companies he
has been involved with and been a key member of the various boards M&A teams achieving over $2 billion in Acquisitions. Mr. Ferrari
continues to mentor and advise a number of CEO’s and start-up companies on strategy and building organizations dedicated to delivering
excellence. Mr. Ferrari is the creator of Excellence by Choice a series of lectures and presentations to help early-stage companies perform
at the highest level of execution. Mr. Ferrari received a Bachelor Degree in Education from Ashland University and a MBA from University
of South Florida.
Ivan Howard is a director of the Company.
Mr. Howard has been since 2019 and currently is a Vice President and Sr. Specialist in Alternative Investment Fiduciary Risk for Banco
Santander, a multinational financial services company. From 2020 Mr. Howard has been and currently serves as Director on the Collier County
Farm Bureau board of directors. From 2016, Mr. Howard has been and currently serves as Chairman of the Hendry/Glades County Farm Service
Agency. From 2020 Mr. Howard has been and currently serves on the U.S. Department of Agriculture Advisory Committee on Minority Farmers.
From 2018 Mr. Howard has been and is currently a member of the University of Florida College of Biomedical Engineering External Advisory
board. Mr. Howard holds an MBA from Mercer University and a Master’s Degree in Biomedical Engineering from the University of Florida.
We believe that Mr. Howard is well qualified to
serve as a Director on our Board with his financial services and board membership experience.
Kristine M. Jacques was appointed as a
director of the Company on March 25, 2024. From 2017 until 2023, Ms. Jacques was Vice President and General Manager, Interventional Pain
Therapies at Vivex Biologics, Inc., a medical device company where she implemented a comprehensive strategic plan of a disruptive technology
in the interventional spine market serving a significant unmet clinical need and potential $38 billion plus total addressable market,
non-surgical treatment for chronic low back pain. From 2007 to 2017 Ms. Jacques was a Vice President at Alphatec Spine, Inc (Nasdaq:ATEC),
a medical device company where she led the development and execution of a 3-year portfolio strategy to grow market share through identifying
opportunities for innovation, maximizing product positioning and differentiation and delivering high quality products to meet the clinical
and unmet needs of surgeons and their patients. From 1995 until 2007, Ms. Jacques served in various management positions at General Electric
Corporation, prior to which she served from 1991 until 1994 at various management positions at Smith & Nephew, PLC, both of which
are publicly traded. Previously, she was an Account Manager, Senior Investment Analyst for General Electric Capital Corporation from 1988
until 1991. Ms. Jacques received a Bachelor of Arts degree in Finance Administration from Michigan State University.
We believe that Ms. Jacques is well qualified
to serve as a Director on our Board with her experience as a senior executive in the spine and medical device industries.
Robert K. Weigle currently is and has been
since October 2020, the CEO of Prime Genomics, a saliva-based diagnostics company utilizing Genomics. Mr Weigle is also currently an executive
in residence with DigitalDX, a venture capital firm. Mr. Weigle was CEO and a director of Benvenue Medical from May 2009 until August
2020. Benvenue was a Silicon Valley based medical device company, which raised over $200 million in funding. At Benvenue Mr. Weigle led
growth from pre-clinical to successful clinical trials to commercial launch of first-generation devices in two distinct markets, one for
the treatment of compression fractures in the spine and the second for the treatment of degenerative disc disease, resulting in a first
full-year run rate exceeding $1 million per month. Mr. Weigle oversaw all early aspects of corporate strategy, including defining, communicating
and executing the company’s overall business model; and represented Benvenue to the investment community. Mr. Weigle was also a
senior executive at numerous healthcare/medical device companies, including TherOx, Inc, Cardiac Pathways, Baxter Healthcare and Cardima
Corporation. Mr. Weigle also has relevant experience at Johnson & Johnson. Mr. Weigle holds a BA in Political Science from University
of California, Berkeley.
We believe that Mr. Weigle is well qualified to
serve as a Director on our Board with his experience in leading medical device companies both as a senior executive and as a member of
the board of directors.
Stephen H. Hochschuler, M.D. is a world-renowned
orthopedic spine surgeon. Dr. Hochschuler is the co-founder of the Texas Back Institute and founder of Back Systems, Inc., and founding
Chairman of Innovative Spinal Technologies, Dr. Hochschuler has severed on numerous boards of directors and advisory boards for medical
and scientific institutions. Dr. Hochschuler is a member of numerous national and international professional organizations including the
American Academy of Orthopedic Surgeons; the American Pain Society; North American Spine Society; and the Southwest Chapter of the Society
of International Business Fellows. Internationally, he is a member of the International Intradiscal Therapy Society; the International
Society for Minimal Intervention in Spinal Surgery; the International Society for the Study of the Lumbar Spine; and is a founding board
member of the Spinal Arthroplasty Society. He has also been a founding board member of The American Board of Spine Surgery and The American
College of Spine Surgery. He is published in a wide range of professional journals, and has delivered numerous presentations worldwide.
Dr. Hochschuler holds a BA from Columbia College and his medical degree from Harvard Medical School.
We believe that Dr. Hochschuler is well qualified
to serve as a Director on our Board with his experience in as an orthopedic spine surgeon and his service on boards of directors and advisory
boards of medical and scientific institutions as a member of the board of directors.
Board Composition
Our business and affairs are managed under the
direction of our Board. Our Board currently consists of seven members, four of whom qualify as “independent” under the listing
standards of Nasdaq.
Directors serve until the next annual meeting
and until their successors are elected and qualified. Officers are appointed to serve for one year until the meeting of the Board following
the annual meeting of shareholders and until their successors have been elected and qualified.
Director Independence
Our Board is composed of a majority of “independent
directors” as defined under the rules of Nasdaq. We use the definition of “independence” applied by Nasdaq to
make this determination. Nasdaq Listing Rule 5605(a)(2) provides that an “independent director” is a person other than
an officer or employee of the company or any other individual having a relationship which, in the opinion of the Board, would interfere
with the exercise of independent judgment in carrying out the responsibilities of a director. The Nasdaq listing rules provide that a
director cannot be considered independent if:
| |
● |
the director is, or at any time during the past three (3) years was, an employee of the company; |
| |
|
|
| |
● |
the director or a family member of the director accepted any compensation from the company in excess of $120,000 during any period of twelve (12) consecutive months within the three (3) years preceding the independence determination (subject to certain exemptions, including, among other things, compensation for board or board committee service); |
| |
|
|
| |
● |
the director or a family member of the director is a partner in, controlling shareholder of, or an executive officer of an entity to which the company made, or from which the company received, payments in the current or any of the past three fiscal years that exceed 5% of the recipient’s consolidated gross revenue for that year or $200,000, whichever is greater (subject to certain exemptions); |
| |
|
|
| |
● |
the director or a family member of the director is employed as an executive officer of an entity where, at any time during the past three (3) years, any of the executive officers of the company served on the compensation committee of such other entity; or |
| |
|
|
| |
● |
the director or a family member of the director is a current partner of the company’s outside auditor, or at any time during the past three (3) years was a partner or employee of the company’s outside auditor, and who worked on the company’s audit. |
Under such definitions, our Board has undertaken
a review of the independence of each director. Based on the information provided by each director concerning his or her background, employment,
and affiliations, our Board has determined that Ivan Howard, Robert K. Weigle, Stephen H. Hochschuler, M.D. and Kristine Jacques are independent
directors of the Company.
Board Committees
The Board has established three standing committees:
(i) audit committee (the “Audit Committee”); (ii) compensation committee (the “Compensation Committee”); and (iii)
nominating and corporate governance committee (the “Nominating and Corporate Governance Committee”). Each of the committees
operates pursuant to its charter. The committee charters will be reviewed annually by the Nominating and Corporate Governance Committee.
If appropriate, and in consultation with the chairs of the other committees, the Nominating and Corporate Governance Committee may propose
revisions to the charters. The responsibilities of each committee are described in more detail below.
Audit Committee. The Audit Committee
consists of three directors, Ivan Howard, Robert Weigle and Kristine Jacques, all of which are currently “independent” as
defined by Nasdaq and includes an audit committee financial expert, Mr. Howard, within the meaning of Item 407(d) of Regulation S-K under
the Securities Act of 1933, as amended, or the Securities Act. The audit committee’s duties are specified in a charter and include,
but not be limited to:
| |
● |
reviewing and discussing with management and the independent auditor the annual audited financial statements, and recommending to the board whether the audited financial statements should be included in our annual disclosure report; |
| |
|
|
| |
● |
discussing with management and the independent auditor significant financial reporting issues and judgments made in connection with the preparation of our financial statements; |
| |
|
|
| |
● |
discussing with management major risk assessment and risk management policies; |
| |
● |
monitoring the independence of the independent auditor; |
| |
|
|
| |
● |
verifying the rotation of the lead (or coordinating) audit partner having primary responsibility for the audit and the audit partner responsible for reviewing the audit as required by law; |
| |
|
|
| |
● |
reviewing and approving all related-party transactions; |
| |
|
|
| |
● |
inquiring and discussing with management our compliance with applicable laws and regulations; |
| |
|
|
| |
● |
pre-approving all audit services and permitted non-audit services to be performed by our independent auditor, including the fees and terms of the services to be performed; |
| |
|
|
| |
● |
appointing or replacing the independent auditor; |
| |
|
|
| |
● |
determining the compensation and oversight of the work of the independent auditor (including resolution of disagreements between management and the independent auditor regarding financial reporting) for the purpose of preparing or issuing an audit report or related work; |
| |
|
|
| |
● |
establishing procedures for the receipt, retention and treatment of complaints received by us regarding accounting, internal accounting controls or reports which raise material issues regarding our financial statements or accounting policies; and |
| |
|
|
| |
● |
approving reimbursement of expenses incurred by our management team in identifying potential target businesses. |
The Audit Committee is composed exclusively of
“independent directors” who are “financially literate” as defined under the Nasdaq listing standards. The Nasdaq
listing standards define “financially literate” as being able to read and understand fundamental financial statements, including
a company’s balance sheet, income statement and cash flow statement.
Compensation Committee. The Compensation
Committee consists of three directors, Ivan Howard, Robert Weigle and Kristine Jacques, who are “independent” as defined by
Nasdaq. The Compensation Committee’s duties are specified in a charter and include, but not be limited to:
| |
● |
reviews, approves and determines, or makes recommendations to our Board regarding, the compensation of our executive officers; |
| |
|
|
| |
● |
administers our equity compensation plans; |
| |
|
|
| |
● |
reviews and approves, or makes recommendations to our Board, regarding incentive compensation and equity compensation plans; and |
| |
|
|
| |
● |
establishes and reviews general policies relating to compensation and benefits of our employees. |
Nominating and Corporate Governance Committee.
The Nominating and Corporate Governance Committee consists of two directors, Robert Weigle and Stephen Hochschuler, both of which
are “independent” as defined by Nasdaq. The nominating and corporate governance committee’s duties are specified in
a charter and include, but not be limited to:
| |
● |
identifying, reviewing and evaluating candidates to serve on our Board consistent with criteria approved by our Board; |
| |
|
|
| |
● |
evaluating director performance on our Board and applicable committees of our Board and determining whether continued service on our Board is appropriate; |
| |
|
|
| |
● |
evaluating nominations by stockholders of candidates for election to our Board; and |
| |
|
|
| |
● |
corporate governance matters. |
Role of Board in Risk Oversight Process
Our Board has responsibility for the oversight
of our risk management processes and, either as a whole or through its committees, regularly discusses with management our major risk
exposures, their potential impact on our business and the steps we take to manage them. The risk oversight process includes receiving
regular reports from board committees and members of senior management to enable our Board to understand our risk identification, risk
management, and risk mitigation strategies with respect to areas of potential material risk, including operations, finance, legal, regulatory,
cybersecurity, strategic and reputational risk.
Code of Ethics
Our Board adopted a written code of business conduct
and ethics (“Code”) that applies to our directors, officers and employees, including our principal executive officer, principal
financial officer and principal accounting officer or controller, or persons performing similar functions. Our website has a current copy
of the Code and all disclosures that are required by law in regard to any amendments to, or waivers from, any provision of the Code.
Family Relationships
There are no family relationships among any of
our executive officers or directors.
Involvement in Certain Legal Proceedings
To our knowledge, none of our current directors
or executive officers has, during the past ten (10) years:
| |
● |
been convicted in a criminal proceeding or been subject to a pending criminal proceeding (excluding traffic violations and other minor offenses); |
| |
|
|
| |
● |
had any bankruptcy petition filed by or against the business or property of the person, or of any partnership, corporation or business association of which he was a general partner or executive officer, either at the time of the bankruptcy filing or within two (2) years prior to that time; |
| |
|
|
| |
● |
been subject to any order, judgment, or decree, not subsequently reversed, suspended or vacated, of any court of competent jurisdiction or federal or state authority, permanently or temporarily enjoining, barring, suspending or otherwise limiting, his involvement in any type of business, securities, futures, commodities, investment, banking, savings and loan or insurance activities, or to be associated with persons engaged in any such activity; |
| |
|
|
| |
● |
been found by a court of competent jurisdiction in a civil action or by the SEC or the Commodity Futures Trading Commission to have violated a federal or state securities or commodities law, and the judgment has not been reversed, suspended or vacated; |
| |
|
|
| |
● |
been the subject of, or a party to, any federal or state judicial or administrative order, judgment, decree or finding, not subsequently reversed, suspended or vacated (not including any settlement of a civil proceeding among private litigants), relating to an alleged violation of any federal or state securities or commodities law or regulation, any law or regulation respecting financial institutions or insurance companies including, but not limited to, a temporary or permanent injunction, order of disgorgement or restitution, civil money penalty or temporary or permanent cease-and-desist order, or removal or prohibition order, or any law or regulation prohibiting mail or wire fraud or fraud in connection with any business entity; or |
| |
|
|
| |
● |
been the subject of, or a party to, any sanction or order, not subsequently reversed, suspended or vacated, of any self-regulatory organization (as defined in Section 3(a)(26) of the Exchange Act), any registered entity (as defined in Section 1(a)(29) of the Commodity Exchange Act), or any equivalent exchange, association, entity or organization that has disciplinary authority over its members or persons associated with a member. |
EXECUTIVE COMPENSATION
The following summary compensation table provides
information regarding the compensation paid during our fiscal years ended December 31, 2023 and December 31, 2022 to our Chief Executive
Officer (principal executive officer), our Chief Financial Officer and Chief Technology Officer. We refer to these individuals as our
“named executive officers.”
Summary Compensation Table
| Name and Principal Position | |
(Salary $) | | |
($) Bonus | | |
Option/RSU Awards(1) ($) | | |
Total ($) | |
| Steven M. Foster, Chief Executive Officer | |
| | |
| | |
| | |
| |
| 2023 | |
$ | 400,000 | | |
$ | 87,600 | | |
$ | - | | |
$ | 487,600 | |
| 2022 | |
$ | 300,000 | | |
$ | 70,000 | | |
$ | 1,926,634 | | |
$ | 2,296,634 | |
| Steven Van Dick, Former Chief Financial Officer(2) | |
| | | |
| | | |
| | | |
| | |
| 2023 | |
$ | 325,000 | | |
$ | 60,225 | | |
$ | - | | |
$ | 385,225 | |
| 2022 | |
$ | 275,000 | | |
$ | 148,125 | | |
$ | 808,998 | | |
$ | 1,232,123 | |
| Richard Ginn, Chief Technology Officer | |
| | | |
| | | |
| | | |
| | |
| 2023 | |
$ | 290,000 | | |
$ | 60,225 | | |
$ | - | | |
$ | 350,225 | |
| 2022 | |
$ | 275,000 | | |
$ | 148,125 | | |
$ | 3,995,603 | | |
$ | 4,418,728 | |
| (1) | In
2022 the named executives received restricted stock units (“RSUs”). |
| | |
| (2) | Retired on July 31, 2024. |
Employment
Agreements
We have executed the following employment agreements
with our named executive officers. The material terms of each of those arrangements are summarized below. The summaries are not a complete
description of all provisions of the employment arrangements and are qualified in their entirety by reference to the written employment
arrangements, each filed as an exhibit to the registration statement of which this prospectus is a part.
Foster Employment Agreement.
Steven M. Foster, our Chief Executive Officer and President and a member of our Board, and the Company entered into an Employment Agreement
dated as of June 1, 2021 (the “Foster Employment Agreement”). The Foster Employment Agreement provides Mr. Foster an annual
base salary of $300,000, an annual bonus of up to $120,000 based upon achievement of mutually agreed upon milestones, options to purchase
shares of our common stock in an amount sufficient to maintain Mr. Foster’s equity ownership at 4%, which were granted at the closing
of our initial public offering and employee benefits that are generally given to our senior executives.
Under the Foster Employment Agreement, in the
event that Mr. Foster’s employment is terminated by us without cause (as described in the Foster Employment Agreement) or by Mr.
Foster for good reason (as described in the Foster Employment Agreement), Mr. Foster would be entitled to (1) severance equal to his base
salary at termination, payable in instalments over the 12-month period following termination and (2) payments in respect of continuing
health care coverage for up to twelve months following termination. In addition, upon a change in control of the Company, Mr. Foster would
be entitled to (1) vesting of his options granted prior to the date of the Foster Employment Agreement and (2) a lump sum cash payment
of one year of his base salary and bonus opportunity then in effect.
If Mr. Foster is terminated for cause or because
of death or disability or resigns without good reason, then all vesting of Mr. Foster’s equity awards and payments of compensation
will immediately terminate and any severance benefits will be paid in accordance with established policies, if any, then in effect.
The Foster Employment Agreement contains restrictive
covenants and other obligations relating to non-solicitation of our employees, non-disclosure of our proprietary information and assignment
of inventions.
Ginn Employment Agreement. Richard Ginn,
our founder, Chief Technology Officer and a director of the Company, and the Company entered into an Employment Agreement dated as of
June 1, 2021 (the “Ginn Employment Agreement”). The Ginn Employment Agreement provides Mr. Ginn an annual base salary of $275,000,
an annual bonus of up to 30% of base salary based upon achievement of mutually agreed upon milestones, a second bonus of up to $200,000
based on certain milestones determined by our Board and employee benefits that are generally given to our senior executives.
Under the Ginn Employment Agreement, in the event
that Mr. Ginn’s employment is terminated by us without cause (as described in the Ginn Employment Agreement) or by Mr. Ginn for
good reason (as described in the Foster Employment Agreement), Mr. Ginn would be entitled to (1) severance equal to his base salary at
termination, payable in instalments over the 12-month period following termination and (2) payments in respect of continuing health care
coverage for up to twelve months following termination. In addition, upon a change in control of the Company, Mr. Ginn would be entitled
to (1) vesting of his options granted prior to the date of the Ginn Employment Agreement and (2) a lump sum cash payment of one year of
his base salary and bonus opportunity.
If Mr. Ginn is terminated for cause or because
of death or disability or resigns without good reason, then all vesting of Mr. Ginn’s equity awards and payments of compensation
will immediately terminate and any severance benefits will be paid in accordance with established policies, if any, then in effect.
The Ginn Employment Agreement contains restrictive
covenants and other obligations relating to non-solicitation of our employees, non-disclosure of our proprietary information and assignment
of inventions.
Van Dick Employment Agreement.
Steven Van Dick, our former Executive Vice President, Finance and Administration and Chief Financial Officer, and the Company entered
into that certain Employment Agreement dated as of June 1, 2021 (the “Van Dick Employment Agreement”). Prior to his retirement
on July 31, 2024, the Van Dick Employment Agreement provided Mr. Van Dick an annual base salary of $275,000, an annual bonus of up to
30% of base salary based upon achievement of mutually agreed upon milestones and employee benefits that are generally given to our senior
executives.
The above summary description of the named
executives’ employment agreement includes some of the general terms and provisions of those agreements. For a more detailed description
of those employment agreements, you should refer to such agreements, which are included as exhibits to the registration statement of which
this prospectus forms a part.
Outstanding Equity Awards at Fiscal Year-End
The following table summarizes the number of RSUs
and shares of common stock underlying outstanding equity incentive plan awards for each named executive officer as of December 31, 2023.
| | |
Option Awards | |
Equity Awards (RSUs) | |
| Name | |
Number of
Securities
Underlying
Unexercised
Options (#)
Exercisable | | |
Number of
Securities
Underlying
Unexercised
Options (#)
Unexercisable | | |
Option Exercise
Price ($) | | |
Option Expiration
Date | |
Number of RSUs
that have not Vested | | |
Market
Value of
RSUs | |
| Steven M. Foster(1) | |
| 9,687 | | |
| 1,563 | | |
$ | 52.00 | | |
May 1, 2031 | |
| 10,874 | | |
$ | 17,181 | |
| Steven Van Dick(2) | |
| 4,865 | | |
| 785 | | |
$ | 52.00 | | |
May 1, 2031 | |
| 4,566 | | |
$ | 7,214 | |
| | |
| 2,786 | | |
| 673 | | |
$ | 70.60 | | |
July 19, 2031 | |
| | | |
| | |
| Richard Ginn(3) | |
| 4,865 | | |
| 785 | | |
$ | 52.00 | | |
May 1, 2031 | |
| 22,551 | | |
$ | 35,631 | |
| | |
| 443 | | |
| 107 | | |
$ | 70.60 | | |
July 19, 2031 | |
| | | |
| | |
| (1) | 11,250
option shares were exchanged for 5,625 restricted stock units on May 6, 2024. |
| (2) | 9,109
option shares were exchanged for 4,555 restricted stock units on May 6, 2024. Retired on July 31, 2024. |
| (3) | 6,200
option shares were exchanged for 3,100 restricted stock units on May 6, 2024. |
Stock Options
We granted Steven M. Foster (i) an option to purchase
11,250 shares of common stock at an exercise price of $52.00 per share with a grant date of May 1, 2021, subject to monthly equal vesting
over a three-year period and adjustment in certain circumstances as provided therein (9,687 shares of which are vested), and (ii) a restricted
stock unit consisting of 21,746 shares of common stock with a grant date of May 12, 2022, subject to semi-annual vesting over a three-year
period commencing May 22, 2022, with a one-year cliff.
We granted Steven Van Dick (i) an option to purchase
5,650 shares of common stock at an exercise of $52.00 per share with a grant date of May 1, 2021, subject to monthly equal vesting over
a three-year period that commenced on November 1, 2020 (4,865 shares of which are vested), (ii) an option to purchase 3,459 shares of
common stock at an exercise price of $70.60 per share with a grant date of July 19, 2021, subject to monthly equal vesting over a three-year
period commencing July 19, 2021 (2,786 shares of which are vested), and (iii) a restricted stock unit consisting of 9,131 shares of common
stock with a grant date of May 12, 2022, subject to semi-annual vesting over a three-year period commencing May 22, 2022, with a one-year
cliff.
We granted Richard Ginn (i) an option to purchase
5,650 shares of common stock at an exercise price of $52.00 per share with a grant date of May 1, 2021, subject to monthly equal vesting
over a three-year period commencing April 1, 2021 (4,865 shares of which are vested), (ii) an option to purchase 550 shares of common
stock at an exercise price of $70.60 per share with a grant date of July 19, 2021, subject to monthly equal vesting over a three-year
period commencing July 19, 2021 (443 shares of which are vested) and (iii) a restricted stock unit consisting of 45,098 shares of common
stock with a grant date of May 12, 2022, subject to semi-annual vesting over a three-year period commencing May 22, 2022, with a one-year
cliff.
RSUs
All of the RSUs were either granted on May 12,
2022 and have the following vesting schedule: one-third vest on May 22, 2023 and the remaining two thirds vesting equally every six months
over the following two years, or on May 6, 2024 pursuant to the Option Exchange.
Board Compensation
The following summary board compensation table
provides information regarding the board compensation paid during our fiscal year ended December 31, 2023 to our board members. Only our
independent directors received compensation for being directors during fiscal year 2023.
| Director | |
Cash
Compensation1 | | |
Equity
Compensation2 | | |
Total
Compensation | |
| Frank Fischer | |
$ | 60,000 | | |
$ | - | | |
$ | 60,000 | |
| Ivan Howard | |
$ | 60,000 | | |
$ | - | | |
$ | 60,000 | |
| Kristine M. Jacques3 | |
$ | - | | |
$ | - | | |
$ | - | |
| Robert Weigle | |
$ | 67,500 | | |
$ | - | | |
$ | 67,500 | |
| Stephen Hochschuler | |
$ | 45,000 | | |
$ | - | | |
$ | 45,000 | |
| Total | |
| 232,500 | | |
$ | - | | |
$ | 232,500 | |
| 1 | Frank
Fischer received $40,000 as a board retainer and $20,000 for being Compensation Committee Chairman; Ivan Howard received $40,000 as a
board retainer and $20,000 for being Audit Committee Chairman; Robert Weigle received $40,000 as a board retainer, $10,000 for being
Nominating and Corporate Governance Committee Chairman, $7,500 for being a member of the Compensation Committee and $10,000 for being
a member of the Audit Committee; and Stephen Hochschuler received $40,000 as a board retainer and $5,000 for being a member of the Nominating
and Corporate Governance Committee. |
| 2 | No
equity compensation was issued to board members in 2023. |
| 3 | Appointed
as a director on March 25, 2024. |
Executive Chairman
On May 7, 2021, the Company entered into a Consulting
Agreement (the “Ferrari Consulting Agreement”) with Richard Ferrari, a founder of the Company and its Executive Chairman,
pursuant to which Mr. Ferrari was to assume the role of Executive Chairman of the Company in exchange for compensation of $22,500 per
month starting September 1, 2021. Under this consulting agreement Mr. Ferrari was paid a bonus of $350,000, as a result of the closing
of our initial public offering in April 2022. In May of 2022 Mr. Ferrari was granted RSUs which had a grant date fair value of $2,427,020
and vest over three years, with one-third vesting in May of 2023 and the remaining two thirds vesting equally every six months over the
following two years. The compensation paid to Mr. Ferrari during the fiscal year ended December 31, 2023, totaled $247,500. On May 8,
2024, the Compensation Committee approved a two year extension to the Ferrari Consulting Agreement on the same terms.
2022 Equity Incentive Plan
Overview
On January 10, 2022,
our Board approved the 2022 Plan and on February 2, 2020, our stockholders approved the 2022 Plan. The 2022 Plan governs equity awards
to our employees, directors, officers, consultants and other eligible participants. Initially, the maximum number of shares of our common
stock that may be subject to awards under the 2022 Plan is equal to (i) 160,000 plus (ii) the lesser of (a) 75,000 and (b) the number
of shares of our common stock subject to awards granted under the 2012 Plan that after the 2012 Plan is terminated are cancelled, expired
or otherwise terminated without having been exercised in full, are tendered to or withheld by the Company for payment of an exercise price
or for tax withholding obligations, or are forfeited to or repurchased by the Company due to failure to vest. The maximum number of shares
that are subject to awards under the 2022 is subject to an annual increase equal to the lesser of (i) 110,000 shares of our common stock,
(ii) a number of shares of our common stock equal to 4% of the prior year’s maximum number and (iii) such number of shares of our
common stock as determined by the 2022 Plan administrator.
The purpose of 2022 Plan
is to attract and retain the best available personnel for positions of substantial responsibility, to provide additional incentive to
employees, directors and consultants, and to promote the success of our business. The administrator of the 2022 Plan may, in its sole
discretion, amend, alter, suspend or terminate the 2022 Plan, or any part thereof, at any time and for any reason. We will obtain stockholder
approval of any Plan amendment to the extent necessary and desirable to comply with legal and regulatory requirements relating to the
administration of equity-based awards. Unless earlier terminated by the administrator, the 2022 Plan will terminate ten years from the
date it is adopted by our Board.
Authorized Shares
Initially, the maximum
number of shares of our common stock that may be subject to awards under the 2022 Plan is equal to (i) 160,000 plus (ii) the lesser of
(a) 75,000 and (b) the number of shares of our common stock subject to awards granted under the 2012 Plan that after the 2012 Plan is
terminated are cancelled, expired or otherwise terminated without having been exercised in full, are tendered to or withheld by the Company
for payment of an exercise price or for tax withholding obligations, or are forfeited to or repurchased by the Company due to failure
to vest. The maximum number of shares that are subject to awards under the 2022 is subject to an annual increase equal to the lesser of
(i) 110,000 shares of our common stock, (ii) a number of shares of our common stock equal to 4% of the prior year’s maximum number
and (iii) such number of shares of our common stock as determined by the 2022 Plan administrator.
On July 23, 2024, at
our annual meeting, our stockholders voted to amend the 2022 Plan to increase the number of shares reserved for issuance under the 2022
Plan by 1,100,000 shares. The are currently shares 1,408,959 reserved for issuance under the 2022 Plan, of which 1,154,173 shares are
available for future grants.
Additionally, if any
award issued pursuant to the 2022 Plan expires or becomes unexercisable without having been exercised in full, is surrendered pursuant
to an exchange program, as provided in the 2022 Plan, or, with respect to restricted stock, restricted stock units, performance units
or performance shares, is forfeited to or repurchased by us due to the failure to vest, the unpurchased shares (or for awards other than
stock options or stock appreciation rights the forfeited or repurchased shares) which were subject thereto will become available for future
grant or sale under the 2022 Plan (unless the 2022 Plan has terminated). With respect to stock appreciation rights, only shares actually
issued pursuant to a stock appreciation right will cease to be available under the 2022 Plan; all remaining shares under stock appreciation
rights will remain available for future grant or sale under the 2022 Plan (unless the 2022 Plan has terminated). Shares that have actually
been issued under the 2022 Plan under any award will not be returned to the 2022 Plan and will not become available for future distribution
under the 2022 Plan; provided, however, that if shares issued pursuant to awards of restricted stock, restricted stock units, performance
shares or performance units are repurchased by us or are forfeited to us due to the failure to vest, such shares will become available
for future grant under the 2022 Plan. Shares used to pay the exercise price of an award or to satisfy the tax withholdings related to
an award will become available for future grant or sale under the 2022 Plan. To the extent an award under the 2022 Plan is paid out in
cash rather than shares, such cash payment will not result in reducing the number of shares available for issuance under the 2022 Plan.
Notwithstanding the foregoing and, subject to adjustment as provided in the 2022 Plan, the maximum number of shares that may be issued
upon the exercise of incentive stock options will equal the aggregate share number stated above, plus, to the extent allowable under Section
422 of the Code and regulations promulgated thereunder, any shares that become available for issuance under the 2022 Plan in accordance
with the foregoing.
Plan Administration
One or more committees
appointed by our Board will administer the 2022 Plan. Initially, the Compensation Committee shall administer the 2022 Plan. In addition,
if we determine it is desirable to qualify transactions under the 2022 Plan as exempt under Rule 16b-3 of the Exchange Act, such transactions
will be structured with the intent that they satisfy the requirements for exemption under Rule 16b-3. Subject to the provisions of the
2022 Plan, the administrator has the power to administer the 2022 Plan and make all determinations deemed necessary or advisable for administering
the 2022 Plan, including the power to determine the fair market value of our common stock, select the service providers to whom awards
may be granted, determine the number of shares covered by each award, approve forms of award agreements for use under the 2022 Plan, determine
the terms and conditions of awards (including the exercise price, the time or times at which the awards may be exercised, any vesting
acceleration or waiver or forfeiture restrictions and any restriction or limitation regarding any award or the shares relating thereto),
construe and interpret the terms of the 2022 Plan and awards granted under it, prescribe, amend and rescind rules relating to the 2022
Plan, rules and regulations relating to sub-plans established for the purpose of facilitating compliance with applicable non-U.S. laws,
easing the administration of the 2022 Plan and/or for qualifying for favorable tax treatment under applicable non-U.S. laws, in each case
as the administrator may deem necessary or advisable and modify or amend each award (subject to the provisions of the 2022 Plan), including
the discretionary authority to extend the post-termination exercisability period of awards and to extend the maximum term of an option
or stock appreciation right (subject to the provisions of the 2022 Plan), to allow Participants to satisfy withholding tax obligations
in a manner permissible under the 2022 Plan, to authorize any person to execute on behalf of us any instrument required to effect the
grant of an award previously granted by the administrator and to allow a participant to defer the receipt of payment of cash or the delivery
of shares that would otherwise be due to such participant under an award. The administrator also has the authority to allow participants
the opportunity to transfer outstanding awards to a financial institution or other person or entity selected by the administrator and
to institute an exchange program by which outstanding awards may be surrendered or cancelled in exchange for awards of the same type which
may have a higher or lower exercise price or different terms, awards of a different type or cash, or by which the exercise price of an
outstanding award is increased or reduced. The administrator’s decisions, interpretations and other actions are final and binding
on all participants.
Eligibility
Awards under the 2022
Plan, other than incentive stock options, may be granted to our employees (including officers and directors) or a parent or subsidiary,
members of our Board or consultants engaged to render bona fide services to us or a parent or subsidiary. Incentive stock options may
be granted only to our employees or a subsidiary, provided the services (a) are not in connection with the offer or sale of securities
in a capital-raising transaction, and (b) do not directly promote or maintain a market for our securities, in each case, within the meaning
of Form S-8 promulgated under the Securities Act, and provided further, that a consultant will include only those persons to whom the
issuance of Shares may be registered under Form S-8 promulgated under the Securities Act.
Stock Options
Stock options may be
granted under the 2022 Plan. The exercise price of options granted under the 2022 Plan generally must at least be equal to the fair market
value of our common stock on the date of grant. The term of each option will be as stated in the applicable award agreement; provided,
however, that the term may be no more than 10 years from the date of grant. The administrator will determine the methods of payment of
the exercise price of an option, which may include cash, shares or other property acceptable to the administrator, as well as other types
of consideration permitted by applicable law. After the termination of service of an employee, director or consultant, they may exercise
their option for the period of time stated in their option agreement. In the absence of a specified time in an award agreement, if termination
is due to death or disability, the option will remain exercisable for six months. In all other cases, in the absence of a specified time
in an award agreement, the option will remain exercisable for three months following the termination of service. An option may not be
exercised later than the expiration of its term. Subject to the provisions of the 2022 Plan, the administrator determines the other terms
of options.
Stock Appreciation
Rights
Stock appreciation rights
may be granted under the 2022 Plan. Stock appreciation rights allow the recipient to receive the appreciation in the fair market value
of our common stock between the exercise date and the date of grant. Stock appreciation rights may not have a term exceeding 10 years.
After the termination of service of an employee, director or consultant, they may exercise their stock appreciation right for the period
of time stated in their stock appreciation right agreement. In the absence of a specified time in an award agreement, if termination is
due to death or disability, the stock appreciation rights will remain exercisable for six months. In all other cases, in the absence of
a specified time in an award agreement, the stock appreciation rights will remain exercisable for three months following the termination
of service. However, in no event may a stock appreciation right be exercised later than the expiration of its term. Subject to the provisions
of the 2022 Plan, the administrator determines the other terms of stock appreciation rights, including when such rights become exercisable
and whether to pay any increased appreciation in cash or with shares of our common stock, or a combination thereof, except that the per
share exercise price for the shares to be issued pursuant to the exercise of a stock appreciation right will be no less than 100% of the
fair market value per share on the date of grant.
Restricted Stock
Restricted stock may
be granted under the 2022 Plan. Restricted stock awards are grants of shares of our common stock that vest in accordance with terms and
conditions established by the administrator. The administrator will determine the number of shares of restricted stock granted to any
employee, director or consultant and, subject to the provisions of the 2022 Plan, will determine the terms and conditions of such awards.
The administrator may impose whatever conditions to vesting it determines to be appropriate (for example, the administrator may set restrictions
based on the achievement of specific performance goals or continued service to us); provided, however, that the administrator, in its
sole discretion, may accelerate the time at which any restrictions will lapse or be removed. Recipients of restricted stock awards generally
will have voting and dividend rights with respect to such shares upon grant without regard to vesting, unless the administrator provides
otherwise. Shares of restricted stock that do not vest are subject to our right of repurchase or forfeiture.
Restricted Stock
Units
RSUs may be granted under
the 2022 Plan. RSUs are bookkeeping entries representing an amount equal to the fair market value of one share of our common stock. Subject
to the provisions of the 2022 Plan, the administrator determines the terms and conditions of RSUs, including the vesting criteria and
the form and timing of payment. The administrator may set vesting criteria based upon the achievement of Company-wide, divisional, business
unit or individual goals (including continued employment or service), applicable federal or state securities laws or any other basis determined
by the administrator in its discretion. The administrator, in its sole discretion, may pay earned RSUs in the form of cash, in shares
of our common stock or in some combination thereof. Notwithstanding the foregoing, the administrator, in its sole discretion, may accelerate
the time at which any vesting requirements will be deemed satisfied.
Performance Awards
Performance awards may
be granted under the 2022 Plan. Performance awards are awards that will result in a payment to a participant only if performance goals
established by the administrator are achieved or the awards otherwise vest. The administrator will set objectives or vesting provisions,
that, depending on the extent to which they are met, will determine the value the payout for the performance awards. The administrator
may set vesting criteria based on the achievement of company-wide, divisional, business unit, or individual goals (including, but not
limited to, continued employment or service), or any other basis determined by the administrator in its discretion. Each performance award’s
threshold, target, and maximum payout values are established by the administrator on or before the grant date. After the grant of a performance
award, the administrator, in its sole discretion, may reduce or waive any performance objectives or other vesting provisions for such
performance award. The administrator, in its sole discretion, may pay earned performance awards in the form of cash, in shares, or in
some combination thereof.
Non-Employee Directors
The 2022 Plan provides
that all non-employee directors will be eligible to receive all types of awards (except for incentive stock options) under the 2022 Plan.
The 2022 Plan includes a maximum limit of $500,000 of equity awards that may be granted to a non-employee director in any fiscal year,
increased to $750,000 in connection with his or her initial service. For purposes of this limitation, the value of equity awards is based
on the grant date fair value (determined in accordance with accounting principles generally accepted in the United States). Any equity
awards granted to a person for their services as an employee, or for their services as a consultant (other than as a non-employee director),
will not count for purposes of the limitation. The maximum limit does not reflect the intended size of any potential compensation or equity
awards to the Company’s non-employee directors.
Non-transferability
of Awards
Unless the administrator
provides otherwise, the 2022 Plan generally does not allow for the transfer of awards other than by will or by the laws of descent and
distribution and only the recipient of an award may exercise an award during their lifetime. If the administrator makes an award transferrable,
such award will contain such additional terms and conditions as the administrator deems appropriate.
Certain Adjustments
In the event of certain
changes in the Company’s capitalization, to prevent diminution or enlargement of the benefits or potential benefits available under
the 2022 Plan, the administrator will adjust the number and class of shares that may be delivered under the 2022 Plan or the number, and
price of shares covered by each outstanding award and the numerical share limits set forth in the 2022 Plan.
Dissolution or
Liquidation
In the event of the Company’s
proposed liquidation or dissolution, the administrator will notify participants as soon as practicable and all awards will terminate immediately
prior to the consummation of such proposed transaction.
Merger or Change
in Control
The 2022 Plan provides
that in the event of the Company’s merger with or into another corporation or entity or a “change in control” (as defined
in the 2022 Plan), each outstanding award will be treated as the administrator determines, including, without limitation, that (i) awards
will be assumed, or substantially equivalent awards will be substituted, by the acquiring or succeeding corporation (or an affiliate thereof)
with appropriate adjustments as to the number and kind of shares and prices; (ii) upon written notice to a participant, that the participant’s
awards will terminate upon or immediately prior to the consummation of such merger or change in control; (iii) outstanding awards will
vest and become exercisable, realizable or payable, or restrictions applicable to an award will lapse, in whole or in part, prior to or
upon consummation of such merger or change in control and, to the extent the administrator determines, terminate upon or immediately prior
to the effectiveness of such merger or change in control; (iv) (A) the termination of an award in exchange for an amount of cash or property,
if any, equal to the amount that would have been attained upon the exercise of such award or realization of the participant’s rights
as of the date of the occurrence of the transaction (and, for the avoidance of doubt, if as of the date of the occurrence of the transaction
the administrator determines in good faith that no amount would have been attained upon the exercise of such award or realization of the
participant’s rights, then such award may be terminated by the Company without payment) or (B) the replacement of such award with
other rights or property selected by the administrator in its sole discretion; or (v) any combination of the foregoing. The administrator
will not be obligated to treat all awards, all awards a participant holds, or all awards of the same type, similarly. In the event that
awards (or portion thereof) are not assumed or substituted for in the event of a merger or change in control, the participant will fully
vest in and have the right to exercise all of their outstanding options and stock appreciation rights, including shares as to which such
awards would not otherwise be vested or exercisable, all restrictions on restricted stock and RSUs or performance awards will lapse and,
with respect to awards with performance-based vesting, all performance goals or other vesting criteria will be deemed achieved at 100%
of target levels and all other terms and conditions met, in all cases, unless specifically provided otherwise under the applicable award
agreement or other written agreement between the participant and the Company or any of the Company’s subsidiaries or parents, as
applicable. If an option or stock appreciation right is not assumed or substituted in the event of a merger or change in control, the
administrator will notify the participant in writing or electronically that the option or stock appreciation right will be exercisable
for a period of time determined by the administrator in its sole discretion and the vested option or stock appreciation right will terminate
upon the expiration of such period.
For awards granted to
an outside director, the outside director will fully vest in and have the right to exercise options and/or stock appreciation rights as
to all of the shares underlying such award, including those shares which would not be vested or exercisable, all restrictions on restricted
stock and RSUs will lapse, and, with respect to awards with performance-based vesting, all performance goals or other vesting criteria
will be deemed achieved at one hundred percent (100%) of target levels and all other terms and conditions met, unless specifically provided
otherwise under the applicable award agreement or other written agreement between the participant and the Company or any of its subsidiaries
or parents, as applicable.
Clawback
Awards will be subject
to any Company clawback policy that the Company is required to adopt pursuant to the listing standards of any national securities exchange
or association on which the Company’s securities are listed or as is otherwise required by the Dodd-Frank Wall Street Reform and
Consumer Protection Act or other applicable laws. The administrator also may specify in an award agreement that the participant’s
rights, payments or benefits with respect to an award will be subject to reduction, cancellation, forfeiture or recoupment upon the occurrence
of certain specified events. The administrator may require a participant to forfeit, return or reimburse the Company all or a portion
of the award or shares issued under the award, any amounts paid under the award and any payments or proceeds paid or provided upon disposition
of the shares issued under the award in order to comply with such clawback policy or applicable laws.
Amendment and Termination
The administrator has
the authority to amend, suspend or terminate the 2022 Plan provided such action does not impair the existing rights of any participant.
The 2022 Plan automatically will terminate on January 10, 2032, unless it is terminated sooner.
Equity Compensation
Plan Information
The table below sets forth information as of December 31, 2023.
| Plan Category | |
Number of
securities
to be
issued upon
exercise of
outstanding
options, warrants
and rights | | |
Weighted-average
exercise
price of
outstanding
options, warrants
and rights | | |
Number of
securities
remaining
available for
future issuance
under equity
compensation
plans (excluding
securities
reflected in
column (a)) | |
| | |
(a) | | |
(b) | | |
(c) | |
| Equity compensation plans approved by security holders | |
| 179,005 | | |
$ | 42.54 | | |
| 37,486 | |
| Equity compensation plans not approved by security holders | |
| - | | |
$ | - | | |
| - | |
| Total | |
| 179,005 | | |
$ | 42.54 | | |
| 37,486 | |
Policies and Practices for Granting Certain
Equity Awards
Our policies and practices regarding the granting
of equity awards are carefully designed to ensure compliance with applicable securities laws and to maintain the integrity of our executive
compensation program. The Compensation Committee is responsible for the timing and terms of equity awards to executives and other eligible
employees.
The timing of equity award grants is determined
with consideration to a variety of factors, including but not limited to, the achievement of pre-established performance targets, market
conditions and internal milestones. The Company does not follow a predetermined schedule for the granting of equity awards; instead, each
grant is considered on a case-by-case basis to align with the Company’s strategic objectives and to ensure the competitiveness of
our compensation packages.
In determining the timing and terms of an equity
award, the Board or the Compensation Committee may consider material nonpublic information to ensure that such grants are made in compliance
with applicable laws and regulations. The Board’s or the Compensation Committee’s procedures to prevent the improper use of
material nonpublic information in connection with the granting of equity awards include oversight by legal counsel and, where appropriate,
delaying the grant of equity awards until the public disclosure of such material nonpublic information.
The Company is committed to maintaining transparency
in its executive compensation practices and to making equity awards in a manner that is not influenced by the timing of the disclosure
of material nonpublic information for the purpose of affecting the value of executive compensation. The Company regularly reviews its
policies and practices related to equity awards to ensure they meet the evolving standards of corporate governance and continue to serve
the best interests of the Company and its shareholders.
Option Exchange
On April 8, 2024, we
launched a one-time stock option exchange program (the “Option Exchange”) pursuant to which eligible participants were able
to exchange outstanding stock options for a lesser amount of new restricted stock units (“RSUs”). Our executive officers,
non-employee directors and consultants were eligible to participate in the Option Exchange. Employees, non-employee directors and consultants
received one RSU for every two shares of our common stock underlying the eligible options surrendered. This “exchange ratio”
(2-for-1) was applied on a grant-by-grant basis. The Option Exchange expired on May 6, 2024 at 11:59 p.m., Eastern Time. At that time,
stock options to purchase 83,391 shares of our common stock were surrendered and 41,698 new RSUs were issued under the 2022 Plan.
PRINCIPAL STOCKHOLDERS
The following table sets forth certain information,
as of August 13, 2024, with respect to the holdings of (1) each person who is the beneficial owner of more than 5% of a class of Company
voting stock, (2) each of our directors, (3) each executive officer, and (4) all of our current directors and executive officers as a
group.
Beneficial ownership of a class of voting stock
is determined in accordance with the rules of the SEC and includes any shares of such class of the Company’s voting stock over which
a person exercises sole or shared voting or investment power, or of which a person has a right to acquire ownership at any time within
60 days of August 13, 2024. Except as otherwise indicated, we believe that the persons named in this table have sole voting and investment
power with respect to all shares of voting stock held by them. Applicable percentage ownership in the following table is based on 3,951,767
shares of common stock, issued and outstanding on August 13, 2024 and [*] shares of common stock issued and outstanding after this offering,
plus, for each individual, any common stock that individual has the right to acquire within 60 days of August 13, 2024 (based upon the
assumed sale of [*] shares of common stock in this offering and assuming no exercise of the Common Warrants.
To the best of our knowledge, except as otherwise
indicated, each of the persons named in the table has sole voting and investment power with respect to the shares of our common stock
beneficially owned by such person, except to the extent such power may be shared with a spouse. To our knowledge, none of the shares listed
below are held under a voting trust or similar agreement, except as noted. To our knowledge, there is no arrangement, including any pledge
by any person of securities of the Company, the operation of which may at a subsequent date result in a change in control of the Company.
| | |
Number of Shares
Beneficially Owned | | |
Beneficial Ownership Percentages | |
| Name and Address of Beneficial Owner(1) | |
Common
Stock | | |
Series A
Preferred
Stock(2) | | |
Percent of
Common
Stock | | |
Percent of
Series A
Preferred
Stock | | |
Percent of
Voting
Stock(3) | |
| Officers and Directors | |
| | |
| | |
| | |
| | |
| |
| Steven M. Foster, Chief Executive Officer and President | |
| 16,196 | | |
| - | | |
| * | | |
| - | | |
| * | |
| Richard Ginn, Chief Technology Officer | |
| 80,405 | (4) | |
| - | | |
| 2.1 | % | |
| - | | |
| 1.3 | % |
| Richard Ferrari, Chairman of the Board | |
| 48,421 | (5) | |
| - | | |
| 1.3 | % | |
| - | | |
| * | |
| Ivan Howard, Director | |
| 8,517 | (6) | |
| - | | |
| * | | |
| - | | |
| * | |
| Robert K. Weigle, Director | |
| 1,242 | | |
| - | | |
| * | | |
| - | | |
| * | |
| Stephen H. Hochschuler, M.D., Director | |
| 6,133 | (7) | |
| - | | |
| * | | |
| - | | |
| * | |
| Kristine M. Jacques, Director | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | |
| | |
| | | |
| | | |
| | | |
| | | |
| | |
| Officers and Directors as a Group | |
| 160,915 | (8) | |
| - | | |
| 4.7 | % | |
| - | | |
| 2.8 | % |
| 5%+ Stockholders | |
| | | |
| | | |
| | | |
| | | |
| | |
| Zuhlke Ventures AG | |
| 244,773 | | |
| - | | |
| 6.5 | % | |
| - | | |
| 3.9 | % |
| TMD Wealth Management | |
| 870,237 | (9) | |
| - | | |
| 23.0 | % | |
| - | | |
| 13.7 | % |
| The Beckham-Shufeldt Family Trust | |
| - | | |
| 66,116 | | |
| - | | |
| 25.7 | % | |
| 10.4 | % |
| Ascent Special Ventures LLC | |
| - | | |
| 67,783 | | |
| - | | |
| 26.4 | % | |
| 10.7 | % |
| (1) | The
principal address of the named officers, directors and 5%+ stockholders of the Company is c/o Tenon Medical, Inc., 104 Cooper Court,
Los Gatos, CA 95032. |
| (2) | Entitles
the holder to 10 votes per share and votes with the common as a single class. |
| (3) | Represents
total ownership percentage with respect to all shares of common stock and Series A Preferred Stock, as a single class. |
| (4) | Includes
23 shares of our common stock underlying restricted stock units that vest within 60 days of August 13, 2024. |
| (5) | Consists
of 9,222 shares held by the Ferrari Family Trust for which Richard Ferrari is trustee and 1,354 shares of our common stock underlying
restricted stock units that vest within 60 days of August 13, 2024 (includes 684 shares of our common stock underlying restricted stock
units held by TCTIG, LLC for which Richard Ferrari is the beneficial owner) and 6,592 shares of our common stock held by TCTIG, LLC and
for which Richard Ferrari has voting control. |
| (6) | Consists
of 684 shares of our common stock underlying restricted stock units held by TCTIG, LLC for which Ivan Howard is the beneficial owner,
and 6,592 shares of our common stock, in each case, held by TCTIG, LLC and for which Ivan Howard is either the beneficial owner or has
voting control. |
| (7) | Includes
1,974 shares of our common that are held by SHKH, LLC, an entity for which Stephen H. Hochschuler has a controlling interest. |
| (8) | Includes
2,205 shares of our common stock underlying restricted stock units that vest within 60 days of August 13, 2024. |
| (9) | Consists
of (i) 358,137 shares of our Common Stock issued to individuals and entities that are clients of TMD Wealth Management and for which
TMD Wealth Management has sole or shared power of disposition and (ii) 512,100 share of our common stock underlying warrants issued to
individuals and entities that are clients of TMD Wealth Management that may be exercised within 60 days of August 13, 2024 and TMD Wealth
Management has sole or shared power to dispose of the shares issued as result of any such exercise. |
CERTAIN RELATIONSHIPS AND RELATED PARTY
TRANSACTIONS
On May 7, 2021 the Company entered into the “Ferrari
Consulting Agreement with Richard Ferrari, a founder of the Company and its Executive Chairman. See “Executive Compensation-Board
Compensation” for a summary description of the terms of the Ferrari Consulting Agreement.
DESCRIPTION
OF SECURITIES
The following summary description sets forth
some of the general terms and provisions of our capital stock. Because this is a summary description, it does not contain all of the information
that may be important to you. For a more detailed description of our capital stock, you should refer to the applicable provisions of the
General Corporation Law of the State of Delaware (the “DGCL”), our charter and our bylaws as currently in effect. Copies of
our amended and restated certificate of incorporation, as amended, and our bylaws are included as exhibits to the registration statement
of which this prospectus forms a part.
General
The total number of shares of stock which the
Company is authorized to issue is 150,000,000 shares of capital stock, consisting of 130,000,000 shares of common stock, $0.001 par value
per share, and 20,000,000 shares of preferred stock, $0.001 par value per share. As of August 13, 2024, there were 3,951,767 shares of
common stock issued and outstanding.
Common Stock
The holders of our common stock are entitled to
the following rights:
Voting Rights. Each share of our
common stock entitles its holder to one vote per share on all matters to be voted or consented upon by the stockholders. Holders of our
common stock are not entitled to cumulative voting rights with respect to the election of directors.
Election of Directors. The
holders of our common stock, voting as a separate class, shall be entitled to elect one member of our Board.
Dividend Rights. Subject to limitations
under Delaware law and preferences that may apply to any shares of preferred stock that we may decide to issue in the future, holders
of our common stock are entitled to receive ratably such dividends or other distributions, if any, as may be declared by our Board out
of funds legally available therefor.
Liquidation Rights. In the event
of the liquidation, dissolution or winding up of our business, the holders of our common stock are entitled to share ratably in the assets
available for distribution after the payment of all of our debts and other liabilities, subject to the prior rights of the holders of
our preferred stock.
Other Matters. The holders of our
common stock have no subscription, redemption or conversion privileges. Our common stock does not entitle its holders to preemptive rights.
All of the outstanding shares of our common stock are fully paid and non-assessable. The rights, preferences and privileges of the holders
of our common stock are subject to the rights of the holders of shares of any series of preferred stock which we may issue in the future.
Preferred Stock
Our Board also has the authority to issue up to
20,000,000 shares of preferred stock in one or more classes or series and to fix the designations, powers, preferences, and rights, and
the qualifications, limitations, or restrictions thereof including dividend rights, dividend rates, conversion rights, voting rights,
terms of redemption, redemption prices, liquidation preferences and the number of shares constituting any class or series, without further
vote or action by the stockholders.
The issuance of any additional shares of preferred
stock could adversely affect the rights of the holders of common stock and, therefore, reduce the value of the common stock. It is not
possible to state the actual effect of the issuance of any shares of preferred stock on the rights of holders of the common stock until
the Board determines the specific rights of the holders of the preferred stock; however, these effects may include:
| ● | Restricting
dividends on the common stock; |
| ● | Diluting
the voting power of the common stock; |
| ● | Impairing
the liquidation rights of the common stock; or |
| ● | Delaying
or preventing a change in control of the Company without consent of the stockholders. |
Series A Preferred Stock
The Certificate of Designations, Rights, and Preferences
of the Series A Preferred Stock (the “Certificate of Designation”) was filed in Delaware on February 20, 2024 and contains
the terms of the Series A Preferred Stock. As of August 13, 2024, there were 256,968 shares of Series A Preferred Stock issued and outstanding.
Conversion. The Series A Preferred
Stock is convertible, at any time, at the option of the holder into shares of Common Stock. Each share of Series A Preferred Stock shall
be convertible, at any time after the date of issuance, at the option of the holder thereof (or, upon a Required Conversion (as defined
below), at the option of the Company), into that number of shares of common stock determined by dividing the Stated Value (as defined
below) for such share of Series A Preferred Stock by $[*]. “Stated Value” means for any share of Series A Preferred Stock,
an amount equal to the product of (x) $15.125 multiplied by (y) the sum of 1 plus the product of (A) 0.06 multiplied by (B) a fraction
equal to the number of days that such share of Series A Preferred Stock has been issued divided by 365. On any date that ten out the last
15 daily VWAPs of the Common Stock is 250% higher than the Conversion Price on such date, then the Company will have the right to require
50% of the Preferred Stock to be converted into shares of Common Stock. Additionally, on and after the time on which the Company has $2.25
million in revenues in any single financial quarter, the Company will have the right to require 50% of the Preferred Stock to be converted
into shares of Common Stock (a “Required Conversion”). The Conversion Price is subject to anti-dilution adjustment as the
result of any subdivision, combination of shares or recapitalization, stock dividends, stock splits and similar transactions affecting
the Common Stock.
Dividends. No dividends are payable
on the Series A Preferred Stock
Voting Rights. The Series A Preferred
Stock will vote together with the Common Stock on all matters other than as required by law at a rate of 10 votes for each share of Series
A Preferred Stock Notwithstanding the foregoing, the vote of an individual holder of Series A Preferred Stock (and underlying Common Stock)
shall be capped at 9.99% (or 4.99% if selected by the holder). Currently, the holders of the Series A Preferred Stock are entitled to
2,569,680 votes.
Liquidation. Upon any liquidation
or winding up of the Company (a “Liquidation”), the holders of Series A Preferred Stock will be entitled to receive in preference
to any other class or series of the Company’s equity securities the greater of (i) the Stated Value plus accrued and unpaid dividends
and (ii) what would be paid if the Series A Preferred Stock plus accrued and unpaid dividends had been converted into Common Stock. A
consolidation or merger of the Company or sale or transfer of all or substantially all of its assets, or any transaction which results
in the stockholders of the Company owning less than 50% of the equity or voting power of the surviving entity (excluding the issuance
of Common Stock in any financing transaction unless more than 50% of the Company’s shares are issued to one stockholder or a number
of stockholders who act as a one group) shall be deemed a Liquidation (a “Deemed Liquidation”) with respect to the shares
of Series A Preferred Stock of any holder who opts to have such occurrence treated as a Deemed Liquidation; provided that if the liquidation
preference payable on a Deemed Liquidation is less than 110% of the stated value of the Series A Preferred Stock, the dividend rate on
any accrued and unpaid dividends payable with respect to such Deemed Liquidation will increase to 10%. All liquidation preferences payable
in respect of a Deemed Liquidation will be payable in shares of Common Stock based on the closing price of the Common Stock on the date
of such Deemed Liquidation.
Other Matters. Consent of the majority
of the holders will be required to (i) amend the Certificate of Incorporation or Bylaws of the Company so as to adversely alter the rights,
preferences, privileges of the Series A Preferred Stock, (ii) create any new class of shares pari passu or senior to the Series
A Preferred Stock or increase or decrease the number of authorized shares of Common Stock or preferred stock, (iii) pay or declare any
dividend on Common Stock or other junior securities, or incur indebtedness in any single transaction in excess of $1 million or (iv) redeem,
purchase or otherwise acquire any share or shares of preferred stock or Common Stock (other than (a) the repurchase of shares of Common
Stock pursuant to a written benefit plan or employment or consulting agreement, or (b) the repurchase of any equity securities in connection
with the Company’s right of first offer with respect to those securities contained in any written agreement with the Company).
Warrants Offered in February 2024 Private Placements
Exercisability. The Series A Warrants
are exercisable at any time after their original issuance up to the date that is five years after their original issuance. The Series
A Warrants will be exercisable, at the option of each holder, in whole or in part by delivering to us a duly executed exercise notice
accompanied by payment in full in immediately available funds for the number of shares of our common stock subscribed for upon such exercise
(except in the case of a cashless exercise as discussed below). If a registration statement registering the issuance of the shares of
our common stock underlying the Series A Warrants under the Securities Act is not effective or available, the holder may, in its sole
discretion, elect to exercise the Series A Warrants through a cashless exercise, in which case the holder would receive upon such exercise
the net number of shares of our common stock determined according to the formula set forth in the Series A Warrants, as applicable. No
fractional shares of our common stock will be issued in connection with the exercise of a warrant. In lieu of fractional shares, we will
pay the holder an amount in cash equal to the fractional amount multiplied by the exercise price.
Exercise Limitation. A holder will
not have the right to exercise any portion of the Series A Warrants if the holder (together with its affiliates) would beneficially own
in excess of 9.99% of the number of shares of our common stock outstanding immediately after giving effect to the exercise, as such percentage
ownership is determined in accordance with the terms of the Series A Warrants.
Exercise Price. The exercise price
of Series A Warrants is $1.2705 per share. The exercise price and number of shares of common stock issuable upon exercise will adjust
in the event of certain stock dividends and distributions, stock splits, stock combinations, reclassifications, dilutive issuances or
similar events.
Rights as a Shareholder. Except
as otherwise provided in the Series A Warrants or by virtue of such holder’s ownership of our shares of common stock, the holder
of a warrant does not have the rights or privileges of a holder of our shares of common stock, including any voting rights, until the
holder exercises the warrant.
Transferability. Subject to applicable
laws, the Series A Warrants may be offered for sale, sold, transferred or assigned without our consent.
Governing Law. The Series A Warrants
are governed by New York law.
Warrants Offered in November 2023 Private Placement
Exercisability. The Note Warrants
are exercisable at any time after their original issuance up to the date that is five years after their original issuance. The Note Warrants
will be exercisable, at the option of each holder, in whole or in part by delivering to us a duly executed exercise notice accompanied
by payment in full in immediately available funds for the number of shares of our common stock subscribed for upon such exercise (except
in the case of a cashless exercise as discussed below). If a registration statement registering the issuance of the shares of our common
stock underlying the Note Warrants under the Securities Act is not effective or available, the holder may, in its sole discretion, elect
to exercise the Note Warrants through a cashless exercise, in which case the holder would receive upon such exercise the net number of
shares of our common stock determined according to the formula set forth in the Note Warrants, as applicable. No fractional shares of
our common stock will be issued in connection with the exercise of a warrant. In lieu of fractional shares, we will pay the holder an
amount in cash equal to the fractional amount multiplied by the exercise price.
Exercise Limitation. A holder will
not have the right to exercise any portion of the Note Warrants if the holder (together with its affiliates) would beneficially own in
excess of 9.99% of the number of shares of our common stock outstanding immediately after giving effect to the exercise, as such percentage
ownership is determined in accordance with the terms of the Note Warrants.
Exercise Price. The exercise price
of Note Warrants is $1.94 per share. The exercise price and number of shares of common stock issuable upon exercise will adjust in the
event of certain stock dividends and distributions, stock splits, stock combinations, reclassifications, dilutive issuances or similar
events.
Rights as a Shareholder. Except
as otherwise provided in the Note Warrants or by virtue of such holder’s ownership of our shares of common stock, the holder of
a warrant does not have the rights or privileges of a holder of our shares of common stock, including any voting rights, until the holder
exercises the warrant.
Transferability. Subject to applicable
laws, the Note Warrants may be offered for sale, sold, transferred or assigned without our consent.
Governing Law. The Note Warrants
are governed by New York law.
Warrants Offered in June 2023 Public Offering
Exercisability. The Tradeable Warrants
are exercisable at any time after their original issuance up to the date that is five years after their original issuance. The Tradeable
Warrants will be exercisable, at the option of each holder, in whole or in part by delivering to us a duly executed exercise notice accompanied
by payment in full in immediately available funds for the number of shares of our common stock subscribed for upon such exercise (except
in the case of a cashless exercise as discussed below). If a registration statement registering the issuance of the shares of our common
stock underlying the Tradeable Warrants under the Securities Act is not effective or available, the holder may, in its sole discretion,
elect to exercise the Tradeable Warrants through a cashless exercise, in which case the holder would receive upon such exercise the net
number of shares of our common stock determined according to the formula set forth in the Tradeable Warrants, as applicable. No fractional
shares of our common stock will be issued in connection with the exercise of a Warrant. In lieu of fractional shares, we will pay the
holder an amount in cash equal to the fractional amount multiplied by the exercise price.
Exercise Limitation. A holder will
not have the right to exercise any portion of the Tradeable Warrants if the holder (together with its affiliates) would beneficially own
in excess of 4.99% (or, upon election by a holder prior to the issuance of any Tradeable Warrants, 9.99%) of the number of shares of our
common stock outstanding immediately after giving effect to the exercise, as such percentage ownership is determined in accordance with
the terms of the Tradeable Warrants. However, any holder may increase or decrease such percentage to any other percentage not in excess
of 9.99%, upon at least 61 days’ prior notice from the holder to us with respect to any increase in such percentage.
Exercise Price. The exercise price
of Tradeable Warrants is $3.146 per share. The exercise price and number of shares of common stock issuable upon exercise will adjust
in the event of certain stock dividends and distributions, stock splits, stock combinations, reclassifications, dilutive issuances or
similar events.
Fundamental Transactions. In the
event of a fundamental transaction, as described in the Tradeable Warrants, and generally including, with certain exceptions, any reorganization,
recapitalization or reclassification of our shares of common stock, the sale, transfer or other disposition of all or substantially all
of our properties or assets, our consolidation or merger with or into another person, the acquisition of more than 50% of our outstanding
shares of common stock, or any person or group becoming the beneficial owner of 50% of the voting power represented by our outstanding
shares of common stock, the holders of the Tradeable Warrants will be entitled to receive upon exercise thereof the kind and amount of
securities, cash or other property that the holders would have received had they exercised the Tradeable Warrants immediately prior to
such fundamental transaction. Additionally, as more fully described in the Warrant, in the event of certain fundamental transactions,
the holders of the Tradeable Warrants will be entitled to receive consideration in an amount equal to the Black Scholes value of the remaining
unexercised portion of the Tradeable Warrants on the date of consummation of such fundamental transaction.
Rights as a Shareholder. Except
as otherwise provided in the Tradeable Warrants or by virtue of such holder’s ownership of our shares of common stock, the holder
of a Warrant does not have the rights or privileges of a holder of our shares of common stock, including any voting rights, until the
holder exercises the Warrant.
Warrant Agent; Global Certificate.
Pursuant to warrant agent agreement between us and VStock Transfer, LLC, as Warrant agent, the Tradeable Warrants will be issued in book-entry
form and shall initially be represented only by one or more global warrants deposited with the warrant agent, as custodian on behalf of
The Depository Trust Company, or DTC, and registered in the name of Cede & Co., a nominee of DTC, or as otherwise directed by DTC.
Transferability. Subject to applicable
laws, the Tradeable Warrants may be offered for sale, sold, transferred or assigned without our consent.
Exchange Listing. The Tradeable
Warrants are listed on The Nasdaq Capital Market under the symbol “TNONW.”
Governing Law. The Tradeable Warrants
are governed by New York law.
Underwriters’ Warrants
Upon the closing of our initial public offering,
the underwriters were issued five-year warrants to purchase 9,600 shares of our common stock at an exercise price of $50 per share, which
may be exercised at any time.
Options
We have fully vested options to purchase 1,175
shares of our common stock at an exercise price of $70.60 that expire on August 31, 2024, issued and outstanding.
We have fully vested options to purchase 3,421
shares of our common stock at an exercise price of $23.90 that expire on July 5, 2032, issued and outstanding.
We have options to purchase 3,000 shares of our
common stock at an exercise price of $2.00 that expire on November 6, 2033, which fully vest on November 1, 2026, issued and outstanding.
We have options to purchase 64,967 shares of our
common stock at an exercise price of $0.86 that expire on May 8, 2034 issued and outstanding, 62,967 of which will be fully vested on
March 26, 2026 and 2,000 of which will be fully vested on April 16, 2027.
RSUs
Under the 2022 Plan, between May 2022 and May
2024 we granted restricted stock units to 42 individuals to purchase in aggregate 222,414, of which 136,690 shares are fully vested, issued
and outstanding .
From time to time, we expect to continue to issue
options and RSUs under the 2022 Plan to various of our consultants, employees, officers and directors.
On May 6, 2024, we issued a total of 41,698 restricted
stock units to our employees, non-employee directors and consultants in exchange for the cancellation of options to purchase a total of
83,391 shares.
Exclusive Forum
Our Certificate of Incorporation provides that,
unless we consent in writing to the selection of an alternative forum, the Court of Chancery of the State of Delaware shall be the sole
and exclusive forum for (a) any derivative action or proceeding brought on behalf of the Company, (b) any action asserting a claim of
breach of a fiduciary duty owed by any director, officer, employee or agent of the Company to the Company or the Company’s stockholders,
(c) any action asserting a claim arising pursuant to any provision of the Delaware General Corporation Law, our Certificate of Incorporation
or Bylaws, or (d) any action asserting a claim governed by the internal affairs doctrine, in each case subject to said Court of Chancery
having personal jurisdiction over the indispensable parties named as defendants therein. This exclusive forum provision may limit the
ability of our stockholders to bring a claim in a judicial forum that such stockholders find favorable for disputes with us or our directors
or officers, which may discourage lawsuits against us or our directors or officers. Our Certificate of Incorporation also provides that
this choice of forum provision does not apply to claims arising under federal securities laws.
Section 203 of the Delaware General Corporation
Law
We are subject to the provisions of Section 203
of the DGCL regulating corporate takeovers. This statute prevents certain Delaware corporations, under certain circumstances, from engaging
in a “business combination” with:
| |
● |
a stockholder who owns 15% or more of our outstanding voting stock (otherwise known as an “interested stockholder”); |
| |
|
|
| |
● |
an affiliate of an interested stockholder; or |
| |
|
|
| |
● |
an associate of an interested stockholder, for three years following the date that the stockholder became an interested stockholder. |
| |
|
|
A “business combination” includes
a merger or sale of more than 10% of our assets. However, the above provisions of Section 203 do not apply if:
| |
● |
our Board approves the transaction that made the stockholder an “interested stockholder,” prior to the date of the transaction; or |
| |
|
|
| |
● |
after the completion of the transaction that resulted in the stockholder becoming an interested stockholder, that stockholder owned at least 85% of our voting stock outstanding at the time the transaction commenced, other than statutorily excluded shares of common stock. |
Transfer Agent and Registrar
The transfer agent and registrar for our common
stock is VStock Transfer LLC.
Listing
Our common stock and Tradeable Warrants are listed
on The Nasdaq Capital Market under the symbols “TNON” and “TNONW.”
DESCRIPTION OF SECURITIES WE ARE OFFERING
Common Stock
The
material terms and provisions of our common stock are described under the caption “Description of Capital Stock.”
Pre-funded Warrants to be issued in this Offering
The following summary of certain
terms and conditions of the Pre-funded Warrants is not complete and is subject to, and qualified in its entirety by, the provisions of
Pre-funded Warrant, the form of which is filed as an exhibit to the registration statement of which this prospectus forms a part. Prospective
investors should carefully review the terms and provisions of the form of Pre-funded Warrant for a complete description of the terms and
conditions of the Pre-funded Warrants.
General
The term “pre-funded”
refers to the fact that the purchase price of the Pre-funded Warrants in this offering includes almost the entire exercise price that
will be paid under the Pre-funded Warrants, except for a nominal remaining exercise price of $0.0001. The purpose of the Pre-funded Warrants
is to enable investors that may have restrictions on their ability to beneficially own more than 4.99% (or, at the election of the holder,
9.99%) of our outstanding common stock following the consummation of this offering the opportunity to invest capital into the Company
without triggering their ownership restrictions, by receiving Pre-funded Warrants in lieu of shares of our common stock which would result
in such ownership of more than 4.99% (or, at the election of the holder, 9.99%), and receiving the ability to exercise their option to
purchase the shares underlying the Pre-funded Warrants at a nominal price at a later date.
Form
The Pre-funded Warrants will
be issued as individual warrant agreements to the investors. You should review the form of Pre-funded Warrant, filed as an exhibit to
the registration statement of which this prospectus forms a part, for a complete description of the terms and conditions applicable to
the Pre-funded Warrants.
Exercisability
The Pre-funded Warrants are
exercisable at any time after their original issuance. The Pre-funded Warrants will be exercisable, at the option of each holder, in whole
or in part, by delivering to us a duly executed exercise notice accompanied by payment in full in immediately available funds for the
number of shares of our common stock purchased upon such exercise (except in the case of a cashless exercise as described below). A holder
(together with its affiliates) may not exercise any portion of the Pre-funded Warrant to the extent that the holder would own more than
4.99% (or, at the election of the holder, 9.99%) of the outstanding common stock immediately after exercise, except that upon at least
61 days’ prior notice from the holder to us, the holder may increase the amount of ownership of outstanding stock after exercising
the holder’s Pre-funded Warrants up to 9.99% of the number of shares of our common stock outstanding immediately after giving effect
to the exercise, as such percentage ownership is determined in accordance with the terms of the Pre-funded Warrants.
Duration and Exercise Price
The exercise price per whole
share of our common stock purchasable upon the exercise of the Pre-funded Warrants is $0.0001 per share of common stock. The Pre-funded
Warrants will be immediately exercisable and may be exercised at any time until the Pre-funded Warrants are exercised in full.
Cashless Exercise
If, at any time after the
issuance of the Pre-funded Warrants, the holder exercises its pre-funded warrants and a registration statement registering the issuance
of the shares of common stock underlying the Pre-funded Warrants under the Securities Act is not then effective or available (or a prospectus
is not available for the resale of shares of common stock underlying the Pre-funded Warrants), then in lieu of making the cash payment
otherwise contemplated to be made to us upon such exercise in payment of the aggregate exercise price, the holder shall instead receive
upon such exercise (either in whole or in part) only the net number of shares of common stock determined according to a formula set forth
in the Pre-funded Warrants. Notwithstanding anything to the contrary, in the event we do not have or maintain an effective registration
statement, there are no circumstances that would require us to make any cash payments or net cash settle the Pre-funded Warrants to the
holders.
Transferability
Subject to applicable laws,
the Pre-funded Warrants may be offered for sale, sold, transferred or assigned at the option of the holder upon surrender of the Pre-funded
Warrants to us together with the appropriate instruments of transfer.
Exchange Listing
There is no established trading
market for the Pre-funded Warrants and we do not plan on applying to list the Pre-funded Warrants on The Nasdaq Capital Market any other
national securities exchange or any other nationally recognized trading system.
Fundamental Transactions
If, at any time while the
Pre-funded Warrants are outstanding, (1) we consolidate or merge with or into another corporation whether or not the Company is the surviving
corporation, (2) we sell, lease, license, assign, transfer, convey or otherwise dispose of all or substantially all of our assets, or
any of our significant subsidiaries, (3) any purchase offer, tender offer or exchange offer (whether by us or another individual or entity)
is completed pursuant to which holders of our common stock are permitted to sell, tender or exchange their shares for other securities,
cash or property and has been accepted by the holders of 50% or more of our common stock, (4) we consummate a securities purchase agreement
or other business combination with another person or entity whereby such other person or entity acquires more than 50% of our outstanding
common stock, or (5) we effect any reclassification or recapitalization of our common stock or any compulsory exchange pursuant to which
our common stock is converted into or exchanged for other securities, cash or property, or each, a “Fundamental Transaction,”
then upon any subsequent exercise of pre-funded warrants, the holders thereof will have the right to receive the same amount and kind
of securities, cash or property as they would have been entitled to receive upon the occurrence of such Fundamental Transaction if they
had been, immediately prior to such Fundamental Transaction, the holder of the number of shares of common stock then issuable upon exercise
of those pre-funded warrants, and any additional consideration payable as part of the Fundamental Transaction.
Rights as a Stockholder
Except by virtue of such holder’s
ownership of shares of our common stock or as otherwise set forth in the Pre-Funded Warrants, the holder of a Pre-funded Warrant does
not have the rights or privileges of a holder of our common stock, including any voting rights, until the holder exercises the Pre-funded
Warrant.
Common Warrants to be issued in this Offering
The following summary of certain
terms and provisions of the Common Warrants offered hereby is not complete and is subject to, and qualified in its entirety by the provisions
of the form of Common Warrant, which is filed as an exhibit to the registration statement of which this prospectus is a part. Prospective
investors should carefully review the terms and provisions set forth in the form of Common Warrant.
Duration and Exercise Price.
Each Common Warrant offered hereby will have an initial exercise price per share equal to $[*] (equal to 100% of the public offering price
per share). The Common Warrants will be exercisable immediately on the date of issuance. The Common Warrants will expire on the fifth
anniversary of the Initial Exercise Date of the Warrant Stockholder Approval, as applicable. The exercise price and number of shares of
common stock issuable upon exercise is subject to appropriate adjustment in the event of stock dividends, stock splits, reorganizations
or similar events affecting our common stock and the exercise price. A Common Warrant to purchase one share of our common stock will be
issued for every one share of common stock and Pre-funded Warrant purchased in this offering. The Common Warrants will be issued in certificated
form.
Exercisability. The
Common Warrants will be exercisable, at the option of each holder, in whole or in part, by delivering to us a duly executed exercise notice
accompanied by payment in full for the number of shares of our common stock purchased upon such exercise (except in the case of a cashless
exercise as discussed below). A holder (together with its affiliates) may not exercise any portion of the Common Warrant to the extent
that the holder would own more than 4.99% (or, at the election of the purchaser, 9.99%) of the outstanding common stock immediately after
exercise, except that upon notice from the holder to us, the holder may increase or decrease the beneficial ownership limitation up to
9.99% of the number of shares of our common stock outstanding immediately after giving effect to the exercise, as such percentage ownership
is determined in accordance with the terms of the Common Warrants, provided that any increase in such beneficial ownership limitation
shall not be effective until 61 days following notice from the holder to us. No fractional shares of common stock will be issued in connection
with the exercise of a Common Warrant. In lieu of fractional shares, we will round up to the next whole share.
Cashless Exercise.
If, at the time a holder exercises its Common Warrants, a registration statement registering the issuance of the shares of common stock
underlying the Common Warrants under the Securities Act is not then effective or available, then in lieu of making the cash payment otherwise
contemplated to be made to us upon such exercise in payment of the aggregate exercise price, the holder may elect instead to receive upon
such exercise (either in whole or in part) the net number of shares of common stock determined according to a formula set forth in the
Common Warrants.
Transferability. Subject
to applicable laws, the Common Warrants may be transferred at the election of the holder upon surrender of the Common Warrant to the Company
together with the appropriate instruments of transfer.
Exchange Listing. There
is no established public trading market for the Common Warrants, and we do not expect a market to develop. In addition, we do not intend
to list the Common Warrants on any securities exchange or nationally recognized trading system.
Fundamental Transaction.
In the event of a fundamental transaction, as described in the form of Common Warrant, and generally including any reorganization, recapitalization
or reclassification of our common stock, the sale, transfer or other disposition of all or substantially all of our properties or assets,
our consolidation or merger with or into another person, the acquisition of more than 50% of our outstanding common stock, or any person
or group becoming the beneficial owner of 50% of the voting power represented by our outstanding common stock, the holders of the Common
Warrants will be entitled to receive upon exercise of the common warrants the kind and amount of securities, cash or other property that
the holders would have received had they exercised the Common Warrants immediately prior to such fundamental transaction. Notwithstanding
the foregoing, in the event of a fundamental transaction, the holders of the Common Warrants have the right to require us or a successor
entity to redeem the Common Warrants for cash in the amount of the Black-Scholes Value (as defined in each Common Warrant) of the unexercised
portion of the Common Warrants concurrently with or within 30 days following the consummation of a fundamental transaction.
Right as a Stockholder.
Except as otherwise provided in the Common Warrants or by virtue of such holder’s ownership of shares of our common stock, the holders
of the Common Warrants do not have the rights or privileges of holders of our common stock, including any voting rights, until they acquire
shares of our common stock upon exercise of their Common Warrants.
MATERIAL U.S. FEDERAL INCOME TAX CONSEQUENCES
The following discussion is a summary of the material
U.S. federal income tax consequences of the purchase, ownership and disposition of the shares of common stock, Pre-Funded Warrants and
Warrants acquired pursuant to this offering but does not purport to be a complete analysis of all potential tax effects. The effects of
other U.S. federal tax laws, such as estate and gift tax laws, and any applicable state, local or foreign tax laws are not discussed.
This discussion is based on the Code, Treasury Regulations promulgated thereunder, judicial decisions, and published rulings and administrative
pronouncements of the IRS, in effect as of the date of this offering. These authorities may change or be subject to differing interpretations.
Any such change or differing interpretation may be applied retroactively in a manner that could adversely affect a U.S. holder of our
common stock, Pre-Funded Warrants and Warrants. We have not sought and will not seek any rulings from the IRS regarding the matters discussed
below. There can be no assurance the IRS or a court will not take a contrary position regarding the tax consequences of the purchase,
ownership and disposition of our common stock, Pre-Funded Warrants and Warrants.
We assume in this discussion that each holder
holds shares of our common stock, Pre-Funded Warrants and Warrants as “capital assets” within the meaning of Section 1221
of the Code (generally, property held for investment). This discussion does not address all U.S. federal income tax consequences that
may be relevant to a particular holder’s individual circumstances, including the impact of the alternative minimum tax or the unearned
income Medicare contribution tax. In addition, it does not address consequences relevant to holders subject to particular rules, including,
without limitation:
| |
● |
U.S. expatriates and certain former citizens or long-term residents of the United States; |
| |
|
|
| |
● |
persons holding our common stock, Pre-Funded Warrants or Warrants as part of a hedge, straddle or other risk reduction strategy or as part of a conversion transaction or other integrated investment; |
| |
|
|
| |
● |
banks, insurance companies, and other financial institutions; |
| |
|
|
| |
● |
regulated investment companies or real estate investment trusts; |
| |
|
|
| |
● |
brokers, dealers or traders in securities or currencies; |
| |
|
|
| |
● |
controlled foreign corporations, “passive foreign investment companies,” and corporations that accumulate earnings to avoid U.S. federal income tax; |
| |
|
|
| |
● |
partnerships or other entities or arrangements treated as partnerships for U.S. federal income tax purposes (and investors therein); |
| |
|
|
| |
● |
tax-exempt organizations or governmental organizations; |
| |
|
|
| |
● |
persons deemed to sell our common stock, Pre-Funded Warrants or Warrants under the constructive sale provisions of the Code; |
| |
|
|
| |
● |
persons for whom our common stock or Pre-Funded Warrants constitutes “qualified small business stock” within the meaning of Section 1202 of the Code or as “Section 1244 stock” for purposes of Section 1244 of the Code; |
| |
|
|
| |
● |
persons subject to special tax accounting rules as a result of any item of gross income with respect to our common stock, Pre-Funded Warrants or Warrants being taken into account in an “applicable financial statement” (as defined in the Code); |
| ● | persons
who hold or receive our common stock, Pre-Funded Warrants or Warrants pursuant to the exercise of any employee stock option or otherwise
as compensation; |
| |
● |
tax-qualified retirement plans; and |
| |
|
|
| |
● |
“qualified foreign pension funds” as defined in Section 897(l)(2) of the Code and entities all of the interest of which are held by qualified foreign pension funds |
If a partnership (or other entity treated as a
partnership for U.S. federal income tax purposes) holds our common stock, Pre-Funded Warrants or Warrants, the tax treatment of a partner
in the partnership will depend on the status of the partner, the activities of the partnership and certain determinations made at the
partner level. Accordingly, partnerships holding our common stock, Pre-Funded Warrants or Warrants, and the partners in such partnerships
should consult their tax advisors regarding the U.S. federal income tax consequences to them.
THIS DISCUSSION IS FOR INFORMATION PURPOSES
ONLY AND IS NOT INTENDED AS LEGAL OR TAX ADVICE. INVESTORS SHOULD CONSULT THEIR TAX ADVISORS WITH RESPECT TO THE APPLICATION OF THE U.S.
FEDERAL INCOME TAX LAWS TO THEIR PARTICULAR SITUATIONS AS WELL AS ANY TAX CONSEQUENCES OF THE PURCHASE, OWNERSHIP AND DISPOSITION OF OUR
COMMON STOCK, PRE-FUNDED WARRANTS, AND WARRANTS ARISING UNDER THE U.S. FEDERAL ESTATE OR GIFT TAX LAWS OR UNDER THE LAWS OF ANY STATE,
LOCAL OR NON-U.S. TAXING JURISDICTION OR UNDER ANY APPLICABLE INCOME TAX TREATY.
For purposes of this discussion, a “U.S.
holder” is any beneficial owner of our common stock, Pre-Funded Warrants, or Warrants that, for U.S. federal income tax purposes,
is:
| |
● |
an individual who is a citizen or resident of the United States; |
| |
|
|
| |
● |
a corporation (or other entity treated as a corporation for U.S. federal income tax purposes) created or organized in or under the laws of the United States, any state thereof, or the District of Columbia; |
| |
|
|
| |
● |
an estate, the income of which is subject to U.S. federal income tax regardless of its source; or |
| |
|
|
| |
● |
a trust that (1) is subject to the primary supervision of a U.S. court and the control of one or more United States persons (within the meaning of Section 7701(a)(30) of the Code), or (2) has made a valid election under applicable Treasury Regulations to be treated as a United States person for U.S. federal income tax purposes. |
The term “non-U.S. holder” means any
beneficial owner of our common stock, Pre-funded Warrants or Warrants that is not a U.S. holder and is not a partnership or other entity
properly classified as a partnership for U.S. federal income tax purposes. For the purposes of this discussion, U.S. holders and non-U.S.
holders are referred to collectively as “holders.”
General Treatment of Pre-Funded Warrants
Although it is not entirely free from doubt, a
Pre-Funded Warrant should be treated as a share of our common stock for U.S. federal income tax purposes and a holder of Pre-Funded Warrants
should generally be taxed in the same manner as a holder of common stock as described below. Each holder should consult his, her or its
own tax advisor regarding the risks associated with the acquisition of a Pre-funded Warrant pursuant to this offering (including potential
alternative characterizations). The balance of this discussion generally assumes that the characterization described above is respected
for U.S. federal income tax purposes.
Allocation of Purchase Price Between Share
of Common Stock and Accompanying Warrant to Purchase Our Common Stock
For U.S. federal income tax purposes, each share
of common stock and accompany Common Warrant and Pre-funded Warrant and accompanying Common Warrant should be treated as an “investment
unit” consisting of one share of common stock or one Pre-funded Warrant, as applicable, and a Common Warrant to acquire one share
of our common stock. The purchase price for each investment unit will be allocated between these two components in proportion to their
relative fair market values at the time the unit is purchased by the holder. This allocation of the purchase price for each investment
unit will establish the holder’s initial tax basis for U.S. federal income tax purposes in the share of common stock or Pre-funded
Warrant, as applicable, and the Common Warrant included in each investment unit. The separation of the common stock or Pre-Funded Warrant,
as applicable, and the Warrant included in each investment unit should not be a taxable event for U.S. federal income tax purposes. Each
holder should consult his, her or its own tax advisor regarding the allocation of the purchase price for an investment unit.
U.S. Holders
Exercise or Expiration of Warrants
In general, a U.S. holder will not recognize gain
or loss for U.S. federal income tax purposes upon exercise of a Warrant, except to the extent the U.S. holder receives a cash payment
for any fractional share of common stock that would otherwise have been issuable upon exercise of the Warrant, which will be treated as
a sale subject to the rules described under “- Disposition of Our Common Stock, Pre-Funded Warrants or Warrants” below. The
U.S. holder will take a tax basis in the shares acquired on the exercise of a Warrant equal to the exercise price of the Warrant, increased
by the U.S. holder’s adjusted tax basis in the Warrant exercised (as determined pursuant to the rules discussed above) and decreased
by the adjusted tax basis allocable to any fractional share that would otherwise have been issuable upon exercise of the Warrant. The
U.S. holder’s holding period in the shares of our common stock acquired on exercise of the Warrant will begin on the date of exercise
of the Warrant, and will not include any period for which the U.S. holder held the Warrant.
In certain limited circumstances, a U.S. holder
may be permitted to undertake a cashless exercise of Warrants into our common stock. The U.S. federal income tax treatment of a cashless
exercise of Warrants into our common stock is unclear, and the tax consequences of a cashless exercise could differ from the consequences
upon the exercise of a Warrant described in the preceding paragraph. U.S. holders should consult their own tax advisors regarding the
U.S. federal income tax consequences of a cashless exercise of Warrants.
The lapse or expiration of a Warrant will be treated
as if the U.S. holder sold or exchanged the Warrant and recognized a capital loss equal to the U.S. holder’s tax basis in the Warrant.
The deductibility of capital losses is subject to limitations.
Certain Adjustments to Warrants
Under Section 305 of the Code, an adjustment to
the number of shares of common stock issued on the exercise of the Warrants, or an adjustment to the exercise price of the Warrants, may
be treated as a constructive distribution to a U.S. holder of the Warrants if, and to the extent that, such adjustment has the effect
of increasing such U.S. holder’s proportionate interest in our “earnings and profits” or assets, depending on the circumstances
of such adjustment (for example, if such adjustment is to compensate for a distribution of cash or other property to our shareholders).
An adjustment made pursuant to a bona fide reasonable adjustment formula that has the effect of preventing dilution should generally not
be considered to result in a constructive distribution. Any such constructive distribution would be taxable whether or not there is an
actual distribution of cash or other property to the holders of Warrants. In certain circumstances, if we were to make a distribution
in cash or other property with respect to our common stock after the issuance of the Warrants, then we may make a corresponding distribution
to a Warrant holder. The taxation of a distribution received with respect to a Warrant is unclear. It is possible such a distribution
would be treated as a distribution (or constructive distribution), although other treatments are possible. For more information regarding
the tax considerations related to distributions, see the discussion below regarding “Distributions.” U.S. holders should consult
their tax advisors regarding the proper treatment of any adjustments to the Warrants and any distributions with respect to the Warrants.
Distributions
As described in the section entitled “Dividend
Policy,” we do not anticipate declaring or paying dividends to holders of our common stock in the foreseeable future. However, if
we do make distributions on our common stock or Pre-Funded Warrants to a U.S. holder, such distributions of cash or property generally
will constitute dividends for U.S. federal income tax purposes to the extent paid from our current or accumulated earnings and profits,
as determined under U.S. federal income tax principles. Distributions in excess of our current and accumulated earnings and profits will
constitute a return of capital that is applied against and reduces, but not below zero, a U.S. holder’s adjusted tax basis in our
common stock or Pre-Funded Warrant, as applicable. Any remaining excess will be treated as gain realized on the sale or exchange of our
common stock or Pre-Funded Warrant as described below under the section titled “-Disposition of Our Common Stock, Pre-Funded Warrants
or Warrants.”
Disposition of Our Common Stock, Pre-Funded
Warrants or Warrants
Upon a sale or other taxable disposition of our
common stock, Pre-Funded Warrants or Warrants, a U.S. holder generally will recognize capital gain or loss in an amount equal to the difference
between the amount realized and the U.S. holder’s adjusted tax basis in the common stock, Pre-Funded Warrants or Warrants. Capital
gain or loss will constitute long-term capital gain or loss if the U.S. holder’s holding period for the common stock, Pre-Funded
Warrants or Warrants exceeds one year. The deductibility of capital losses is subject to certain limitations. U.S. holders who recognize
losses with respect to a disposition of our common stock, Pre-Funded Warrants or Warrants should consult their own tax advisors regarding
the tax treatment of such losses.
Information Reporting and Backup Reporting
Information reporting requirements generally will
apply to payments of dividends (including constructive dividends) on the common stock, Pre-Funded Warrants and Warrants and to the proceeds
of a sale or other disposition of common stock, Pre-Funded Warrants and Warrants paid by us to a U.S. holder unless such U.S. holder is
an exempt recipient, such as a corporation. Backup withholding will apply to those payments if the U.S. holder fails to provide the holder’s
taxpayer identification number, or certification of exempt status, or if the holder otherwise fails to comply with applicable requirements
to establish an exemption.
Backup withholding is not an additional tax. Rather,
any amounts withheld under the backup withholding rules will be allowed as a refund or a credit against the U.S. holder’s U.S. federal
income tax liability provided the required information is timely furnished to the IRS. U.S. holders should consult their own tax advisors
regarding their qualification for exemption from information reporting and backup withholding and the procedure for obtaining such exemption.
Non-U.S. Holders
Exercise and Expiration of Warrants
In general, a non-U.S. holder will not recognize
gain or loss for U.S. federal income tax purposes upon the exercise of Warrants into shares of common stock, except to the extent the
non-U.S. holder receives a cash payment for any fractional share of common stock that would otherwise have been issuable upon exercise
of the Warrant, which will be treated as a sale subject to the rules described under “-Disposition of Our Common Stock, Pre-Funded
Warrants or Warrants” below. The U.S. federal income tax treatment of a cashless exercise of Warrants into our common stock is unclear.
A non-U.S. holder should consult his, her, or its own tax advisor regarding the U.S. federal income tax consequences of a cashless exercise
of Warrants.
The expiration of a Warrant will be treated as
if the non-U.S. holder sold or exchanged the Warrant and recognized a capital loss equal to the non-U.S. holder’s tax basis in the
Warrant. However, a non-U.S. holder will not be able to utilize a loss recognized upon expiration of a Warrant against the non-U.S. holder’s
U.S. federal income tax liability unless the loss is effectively connected with the non-U.S. holder’s conduct of a trade or business
within the United States (and, if an income tax treaty applies, is attributable to a permanent establishment or fixed base in the United
States) or is treated as a U.S.-source loss and the non-U.S. holder is present 183 days or more in the taxable year of disposition and
certain other conditions are met.
Certain Adjustments to Warrants
As described under “- U.S. Holders - Certain
Adjustments to Warrants,” an adjustment to the Warrants could result in a constructive distribution to a non-U.S. holder, which
would be treated as described under “Distributions” below. Any resulting withholding tax attributable to deemed dividends
would be collected from other amounts payable or distributable to the non-U.S. holder. Non-U.S. holders should consult their tax advisors
regarding the proper treatment of any adjustments to and distributions on the Warrants.
Distributions
As discussed above, we do not anticipate declaring
or paying dividends in the foreseeable future. However, if we do make distributions on our common stock or Pre-Funded Warrants, such distributions
of cash or property generally will constitute dividends for U.S. federal income tax purposes to the extent paid from our current or accumulated
earnings and profits, as determined under U.S. federal income tax principles. Amounts not treated as dividends for U.S. federal income
tax purposes will constitute a return of capital and first be applied against and reduce a non-U.S. holder’s adjusted tax basis
in its common stock or Pre-Funded Warrants, but not below zero. Any excess will be treated as capital gain and will be treated as described
below in the section relating to the sale or disposition of our common stock, Pre-Funded Warrants or Warrants. Because we may not know
the extent to which a distribution is a dividend for U.S. federal income tax purposes at the time it is made, for purposes of the withholding
rules discussed below we or the applicable withholding agent may treat the entire distribution as a dividend.
Subject to the discussion below on backup withholding
and the Foreign Account Tax Compliance Act, or FACTA,, dividends paid to a non-U.S. holder of our common stock or Pre-Funded Warrants
that are not effectively connected with the non-U.S. holder’s conduct of a trade or business within the United States will
be subject to U.S. federal withholding tax at a rate of 30% of the gross amount of the dividends (or such lower rate specified by an applicable
income tax treaty). In order to receive a reduced treaty rate, you must provide us with an IRS Form W-8BEN, IRS Form W-8BEN-E or other
appropriate version of IRS Form W-8 certifying qualification for the reduced rate.
If dividends paid to a non-U.S. holder are effectively
connected with the non-U.S. holder’s conduct of a trade or business within the United States (and, if required by an applicable
income tax treaty, the non-U.S. holder maintains a permanent establishment in the United States to which such dividends are attributable),
then, although exempt from U.S. federal withholding tax (provided the non-U.S. holder provides appropriate certification, as described
below), the non-U.S. holder will be subject to U.S. federal income tax on such dividends on a net income basis at the regular graduated
rates. In order to obtain this exemption, you must provide us with an IRS Form W-8ECI or other applicable IRS Form W-8 properly certifying
such exemption. In addition, a non-U.S. holder that is a corporation may be subject to a branch profits tax at a rate of 30% (or such
lower rate specified by an applicable income tax treaty) on its effectively connected earnings and profits for the taxable year that are
attributable to such dividends, as adjusted for certain items. Non-U.S. holders should consult their tax advisors regarding their entitlement
to benefits under any applicable income tax treaty and regarding any applicable treaties that may provide for different rules.
If you hold our common stock, Pre-Funded Warrants
or Warrants through a financial institution or other agent acting on your behalf, you will be required to provide appropriate documentation
to the agent, which then will be required to provide certification to us or our paying agent, either directly or through other intermediaries.
You may be eligible to obtain a refund of any excess amounts withheld by timely filing an appropriate claim for refund with the IRS.
Disposition of Our Common Stock, Pre-Funded
Warrants or Warrants
In general, subject to the discussions below on
backup withholding, information reporting and foreign accounts, a non-U.S. holder will not be subject to U.S. federal income tax on any
gain realized upon the sale or other taxable disposition of our common stock, Pre-Funded Warrants or Warrants unless:
| |
● |
the gain is effectively
connected with the non-U.S. holder’s conduct of a trade or business within the United States (and, if required by an applicable
income tax treaty, the non-U.S. holder maintains a permanent establishment or fixed base in the United States to which such gain
is attributable); |
| |
|
|
| |
● |
the non-U.S. holder is
a nonresident alien individual present in the United States for 183 days or more during the taxable year of the disposition and certain
other requirements are met; or |
| |
|
|
| |
● |
our common stock or Pre-Funded
Warrants constitutes U.S. real property interests, or USRPIs, by reason of our status as a U.S. real property holding corporation,
or USRPHC, for U.S. federal income tax purposes at any time within the shorter of the five-year period preceding the non-U.S. holder’s
disposition of, or their holding period for, our common stock, Pre-Funded Warrants or Warrants. |
Gain
described in the first bullet point above will generally be subject to U.S. federal income tax on a net income basis at the regular
rates. A non-U.S. holder that is a corporation also may be subject to a branch profits tax at a rate of 30% (or such lower rate specified
by an applicable income tax treaty) on such effectively connected gain, as adjusted for certain items.
A non-U.S. holder described in the second bullet
point above will be subject to U.S. federal income tax at a rate of 30% (or such lower rate specified by an applicable income tax treaty)
on any gain derived from the disposition, which may be offset by certain U.S. source capital losses of the non-U.S. holder (even though
the individual non-U.S. holder is not considered a resident of the United States) provided the non-U.S. holder has timely filed U.S.
federal income tax returns with respect to such losses.
With respect to the third bullet point above,
we believe we are not currently and do not anticipate becoming a USRPHC. However, because the determination of whether we are a USRPHC
depends on the fair market value of our USRPIs relative to the fair market value of our non-U.S. real property interests and our other
business assets, there can be no assurance we will not become a USRPHC in the future. Even if we are determined to be or were to become
a USRPHC, gain arising from the sale or other taxable disposition by a non-U.S. holder of our common stock, Pre-Funded Warrants or Warrants
will not be subject to U.S. federal income tax if our common stock is “regularly traded,” as defined by applicable Treasury
Regulations, on an established securities market, and such non-U.S. holder owned, actually and constructively, 5% or less of our common
stock throughout the shorter of the five-year period ending on the date of the sale or other taxable disposition or the non-U.S. holder’s
holding period. Special rules may apply to the determination of the 5% threshold in the case of a holder of a Pre-Funded Warrant or Warrant.
Non-U.S. holders are urged to consult their own tax advisors regarding the effect of holding our Pre-Funded Warrants or Warrants on the
calculation of such 5% threshold. If we are a USRPHC and either our common stock is not regularly traded on an established securities
market or a non-U.S. holder holds, or is treated as holding, more than 5% of our outstanding common stock, directly or indirectly, during
the applicable testing period, such non-U.S. holder’s gain on the disposition of shares of our common stock, Pre-Funded or Warrants
generally will be taxed in the same manner as gain that is effectively connected with the conduct of a U.S. trade or business, except
that the branch profits tax generally will not apply. If we are a USRPHC and our common stock is not regularly traded on an established
securities market, a non-U.S. holder’s proceeds received on the disposition of shares will also generally be subject to withholding
at a rate of 15%. No assurance can be provided that our common stock will be regularly traded on an established securities market for
purposes of the rules described above. Prospective investors are encouraged to consult their tax advisors regarding the possible consequences
to them if we are, or were to become, a USRPHC.
Non-U.S. holders should consult their tax advisors
regarding potentially applicable income tax treaties that may provide for different rules.
Information Reporting and Backup Withholding
Generally, we must report annually to the IRS
the amount of distributions (including constructive distributions) on our common stock, Pre-Funded Warrants or Warrants paid to each non-U.S.
holder, their name and address, and the amount of tax withheld, if any. Copies of information returns that are filed with the IRS may
also be made available under the provisions of an applicable treaty or agreement to the tax authorities of the country in which the non-U.S.
holder resides or is established.
Payments of dividends (including constructive
dividends) or of proceeds on the disposition of our common stock, Pre-Funded Warrants or Warrants made to a non-U.S. holder may be subject
to information reporting and backup withholding at a current rate of 24% unless the non-U.S. holder establishes an exemption, for example,
by properly certifying their non-U.S. status on an IRS Form W-8BEN, IRS Form W-8BEN-E or another appropriate version of
IRS Form W-8. Notwithstanding the foregoing, backup withholding and information reporting may apply if either we or our paying agent
has actual knowledge, or reason to know, that a holder is a U.S. person.
Under current U.S. federal income tax law, U.S.
information reporting and backup withholding requirements generally will apply to the proceeds of a disposition of our common stock, Pre-Funded
Warrants or Warrants effected by or through a U.S. office of any broker, U.S. or foreign, except that information reporting and such requirements
may be avoided if the holder provides a properly executed and appropriate IRS Form W-8 or otherwise meets documentary evidence
requirements for establishing non- U.S. holder status or otherwise establishes an exemption. Generally, U.S. information reporting
and backup withholding requirements will not apply to a payment of disposition proceeds to a non-U.S. holder where the transaction is
effected outside the U.S. through a non-U.S. office of a non-U.S. broker. Information reporting and backup withholding requirements may,
however, apply to a payment of disposition proceeds if the broker has actual knowledge, or reason to know, that the holder is, in fact,
a U.S. person. For information reporting purposes, certain brokers with substantial U.S. ownership or operations will generally be treated
in a manner similar to U.S. brokers.
Non-U.S. holders should consult their tax advisors
regarding the application of the information reporting and backup withholding rules to them.
Foreign Account Tax Compliance Act
The Foreign Account Tax Compliance Act and the
rules and regulations promulgated thereunder, collectively FATCA, generally impose withholding tax at a rate of 30% on dividends (including
constructive dividends) on our common stock, Pre-Funded Warrants or Warrants, and certain other withholding payments, if paid to a non-U.S.
entity unless (i) if the non-U.S. entity is a “foreign financial institution,” the non-U.S. entity undertakes certain due
diligence, reporting, withholding, and certification obligations, (ii) if the non-U.S. entity is not a “foreign financial institution,”
the non-U.S. entity identifies certain of its U.S. investors, if any, or (iii) the non-U.S. entity is otherwise exempt under FATCA. While
withholding under FATCA may apply to payments of gross proceeds from a sale or other disposition of our common stock, Pre-Funded Warrants
or Warrants, under proposed U.S. Treasury Regulations withholding on payments of gross proceeds is not required. Although such regulations
are not final, applicable withholding agents may rely on the proposed regulations until final regulations are issued.
The preceding discussion of material U.S. federal
income tax considerations is for informational purposes only. It is not tax advice. Prospective investors should consult their own tax
advisors regarding the particular U.S. federal, state, local and non-U.S. tax consequences of purchasing, holding and disposing of our
common stock, Pre-Funded Warrants or Warrants, including the consequences of any proposed changes in applicable laws.
PLAN OF DISTRIBUTION
[*]. has agreed to act as our sole placement agent
in connection with this offering subject to the terms and conditions of the placement agency agreement dated [*] , 2024. The Placement
Agent is not purchasing or selling any of the securities offered by this prospectus, nor is it required to arrange the purchase or sale
of any specific number or dollar amount of securities but has agreed to use its reasonable best efforts to arrange for the sale of all
of the securities offered hereby. Therefore, we may not sell the entire number of securities offered pursuant to this prospectus. We will
enter into a securities purchase agreement directly with certain investors, at the investor’s option, who purchases our securities
in this offering. Investors who do not enter into a securities purchase agreement shall rely solely on this prospectus in connection with
the purchase of our securities in this offering.
We will deliver the securities being issued to
the investors upon receipt of such investor’s funds for the purchase of the securities offered pursuant to this prospectus. We expect
to deliver the securities being offered pursuant to this prospectus on or about [*] , 2024.
We have agreed to indemnify the Placement Agent
and specified other persons against specified liabilities, including liabilities under the Securities Act, and to contribute to payments
the Placement Agent may be required to make in respect thereof.
Fees and Expenses
We have engaged [*]. as the
sole placement agent in connection with this offering. This offering is being conducted on a “best efforts” basis and the
Placement Agent has no obligation to buy any of the securities from us or to arrange for the purchase or sale of any specific number or
dollar amount of securities. We have agreed to pay the Placement Agent a fee based on the aggregate proceeds as set forth in the table
below:
| | |
Per share
and
Common
Warrant | | |
Per
Pre-funded
Warrant and
Accompanying
Common
Warrant | | |
Total | |
| Public offering price | |
$ | | | |
$ | | | |
$ | | |
| Placement Agent fees(1) | |
$ | | | |
$ | | | |
$ | | |
| Proceeds to us, before expenses(2) | |
$ | | | |
$ | | | |
$ | | |
| (1) |
We have agreed to pay the placement agent a cash fee equal to [*]% of the aggregate purchase price paid by each purchaser of securities that are sold in the offering. We have also agreed to reimburse the placement agent for certain of its offering-related expenses including accountable offering-related legal expenses in an amount up to $[*] and pay the placement agent a non-accountable expense allowance not to exceed $[*]. |
| |
|
| (2) |
The amount of proceeds, before expenses, to us does not give effect to any exercise of the Pre-funded Warrants or Common Warrants. |
We have agreed to pay the
Placement Agent’s accountable legal expenses relating to the offering in the amount of $[*], and to pay the Placement Agent non-accountable
expenses in connection with the offering in the amount of $[*]. We estimate the total expenses payable by us for this offering, excluding
the Placement Agent fees and expenses, will be approximately $ .
The Placement Agent may be
deemed to be an underwriter within the meaning of Section 2(a)(11) of the Securities Act, and any commissions received by it and any profit
realized on the resale of the shares sold by it while acting as principal might be deemed to be underwriting discounts or commissions
under the Securities Act. As an underwriter, the Placement Agent would be required to comply with the requirements of the Securities Act
and the Exchange Act, including, without limitation, Rule 415(a)(4) under the Securities Act and Rule 10b-5 and Regulation M under the
Exchange Act. These rules and regulations may limit the timing of purchases and sales of shares by the Placement Agent acting as principal.
Under these rules and regulations, the Placement Agent:
| |
● |
may not engage in any stabilization activity in connection with our securities; and |
| |
|
|
| |
● |
may not bid for or purchase any of our securities or attempt to induce any person to purchase any of our securities, other than as permitted under the Exchange Act, until it has completed its participation in the distribution. |
Listing
Our common stock is listed on The Nasdaq Capital
Market under the trading symbol “TNON.” We do not plan to list the Pre-funded Warrants or the Common Warrants on the Nasdaq
Capital Market or any other securities exchange or trading market.
Lock-Up Agreements
Pursuant to “lock-up”
agreements, the Company, our executive officers, directors, and certain five percent (5.0%) security holders have agreed, subject to limited
exceptions, not to [*].
Discretionary Accounts
The Placement Agent does
not intend to confirm sales of the securities offered hereby to any accounts over which it has discretionary authority.
Transfer Agent and Registrar
VStock Transfer LLC as the transfer agent and
registrar for our common stock.
Other Activities and Relationships
The Placement Agent and certain of its affiliates
are full service financial institutions engaged in various activities, which may include securities trading, commercial and investment
banking, financial advisory, investment management, investment research, principal investment, hedging, financing and brokerage activities.
The Placement Agent and certain of its affiliates have, from time to time, performed, and may in the future perform, various commercial
and investment banking and financial advisory services for us and our affiliates, for which they received or will receive customary fees
and expenses.
In the ordinary course of their various business
activities, the Placement Agent and certain of its affiliates may make or hold a broad array of investments and actively trade debt and
equity securities (or related derivative securities) and financial instruments (including bank loans) for their own account and for the
accounts of their customers, and such investment and securities activities may involve securities and/or instruments issued by us and
our affiliates. If the Placement Agent or its affiliates have a lending relationship with us, they routinely hedge their credit exposure
to us consistent with their customary risk management policies. The Placement Agent and its affiliates may hedge such exposure by entering
into transactions that consist of either the purchase of credit default swaps or the creation of short positions in our securities or
the securities of our affiliates, including potentially the common stock offered hereby. Any such short position could adversely affect
future trading prices of the common stock offered hereby. The Placement Agent and certain of its affiliates may also communicate independent
investment recommendations, market color or trading ideas and/or publish or express independent research views in respect of such securities
or instruments and may at any time hold, or recommend to clients that they acquire, long and/or short positions in such securities and
instruments.
The foregoing does not purport to be a complete
statement of the terms and conditions of the placement agent agreement or the securities purchase agreement, copies of which are attached
to the registration statement of which this prospectus is a part. See “Where You Can Find More Information.”
EXPERTS
Haskell & White LLP, an independent registered
public accounting firm, audited our consolidated financial statements for the year ended December 31, 2023 and their related audit report
included an explanatory paragraph expressing substantial doubt regarding our ability to continue as a going concern. Armanino LLP, an
independent registered public accounting firm and our former auditor, audited our consolidated financial statements for the year ended
December 31, 2022. We have included our consolidated financial statements with their reports in this prospectus and elsewhere in the registration
statement in reliance on the reports of Haskell & White LLP and Armanino LLP, given on their authority as experts in accounting and
auditing.
CHANGE IN REGISTRANT’S CERTIFYING ACCOUNTANT
On July 28, 2023, we were informed by Armanino
LLP (“Armanino”), our independent registered public accountant prior to September 7, 2023, that Armanino would resign effective
as of the earlier of (i) the date we engaged a new independent registered public accounting firm and (ii) the filing of our Quarterly
Report on Form 10-Q for the fiscal quarter ended September 30, 2023, as a result of Armanino’s determination to cease providing
certain services to public companies. On September 5, 2023, the Audit Committee of the Board of Directors of the Company (the “Audit
Committee”) appointed Haskell & White LLP as the Company’s independent registered public accounting firm for the fiscal
year ending December 31, 2023, effective September 7, 2023 (the “Engagement Date”).
Armanino’s audit reports on the Company’s
consolidated financial statements as of and for the fiscal years ended December 31, 2022 and December 31, 2021 did not contain an adverse
opinion or a disclaimer of opinion, and were not qualified or modified as to uncertainty, audit scope or accounting principles, other
than the explanatory paragraph regarding the Company’s ability to continue as a going concern.
During the fiscal years ended December 31, 2022
and December 31, 2021, and subsequent interim periods through the Engagement Date, there were no disagreements with Armanino on any matter
of accounting principles or practices, financial statement disclosure or auditing scope or procedure, which disagreement(s), if not resolved
to the satisfaction of Armanino, would have caused it to make reference to the subject matter of the disagreement(s) in connection with
its opinion or reportable events under Item 304(a)(1)(v) of Regulation S-K, except that Armanino concurred with the Company’s assessment
of a material weakness related to the Company’s internal controls over financial reporting.
The Audit Committee of
the Company approved the engagement of Haskell & White LLP. During the two most recent fiscal years and through the Engagement Date,
the Company did not consult with Haskell & White LLP regarding either:
| |
1. |
application of accounting principles to any specified transaction, either completed or proposed, or the type of audit opinion that might be rendered on the Company’s financial statements, and neither a written report was provided to the Company nor oral advice was provided that Haskell & White LLP concluded was an important factor considered by the Company in reaching a decision as to the accounting, auditing or financial reporting issue; or |
| |
|
|
| |
2. |
any matter that was either the subject of a disagreement (as defined in Regulation S-K, Item 304(a)(1)(iv) and the related instructions) or reportable event (as defined in Regulation S-K, Item 304(a)(1)(v)). |
In accordance with Item 304(a)(3) of Regulation
S-K, the Company provided Armanino with a copy of the disclosures made herein and requested from Armanino a letter addressed to the Securities
and Exchange Commission indicating whether it agrees with such disclosures. A copy of Armanino’s letter dated as of September 11,
2023 is attached as Exhibit 16.1 hereto.
LEGAL
MATTERS
Certain legal matters with respect to the validity
of the securities being offered by this prospectus will be passed upon by Sichenzia Ross Ference Carmel LLP, New York, New York. [*] is
acting as counsel for the representative of the placement agent with respect to the offering.
WHERE
YOU CAN FIND MORE INFORMATION
We have filed with the
SEC a registration statement on Form S-1 under the Securities Act with respect to the shares of our common stock offered by this prospectus.
This prospectus, which constitutes a part of the registration statement, does not contain all of the information set forth in the registration
statement, some of which is contained in exhibits to the registration statement as permitted by the rules and regulations of the SEC.
For further information with respect to us and our common stock, we refer you to the registration statement, including the exhibits filed
as a part of the registration statement. Statements contained in this prospectus concerning the contents of any contract or any other
document is not necessarily complete. If a contract or document has been filed as an exhibit to the registration statement, please see
the copy of the contract or document that has been filed. Each statement in this prospectus relating to a contract or document filed as
an exhibit is qualified in all respects by the filed exhibit. You may obtain copies of this information by mail from the Public Reference
Section of the SEC, 100 F Street, N.E., Room 1580, Washington, D.C. 20549, at prescribed rates. You may obtain information on the operation
of the public reference rooms by calling the SEC at 1-800-SEC-0330. The SEC also maintains an Internet website that contains reports,
proxy statements and other information about issuers, like us, that file electronically with the SEC. The address of that website is www.sec.gov.
We are subject to the
information and reporting requirements of the Exchange Act and, in accordance with this law, are required to file periodic reports, proxy
statements and other information with the SEC. These periodic reports, proxy statements and other information are available for inspection
and copying at the SEC’s public reference facilities and the website of the SEC referred to above. We also maintain a website at
www.tenonmed.com. You may access these materials free of charge as soon as reasonably practicable after they are electronically
filed with, or furnished to, the SEC. Information contained on our website is not a part of this prospectus and the inclusion of our website
address in this prospectus is an inactive textual reference only.
Tenon Medical, Inc.
Table of Contents to Financial
Statements
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING
FIRM
To the Stockholders and Board of Directors
Tenon Medical, Inc.
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated
balance sheet of Tenon Medical, Inc. (the “Company”) as of December 31, 2023, the related consolidated statements of operations
and comprehensive loss, convertible preferred stock and stockholders’ equity, and cash flows for the year then ended, and the related
notes (collectively, the “consolidated financial statements”). In our opinion, the consolidated financial statements present
fairly, in all material respects, the consolidated financial position of the Company as of December 31, 2023, and the consolidated results
of its operations and its cash flows for the year then ended, in conformity with U.S. generally accepted accounting principles.
Going Concern
The accompanying consolidated financial statements
have been prepared assuming that the Company will continue as a going concern. As described in Note 2 to the consolidated financial statements,
the Company has experienced recurring losses, negative cash flows from operations, and has limited capital resources. These matters raise
substantial doubt about the Company’s ability to continue as a going concern. Management’s plans regarding these matters are
also described in Note 2. The consolidated financial statements do not include any adjustments that might result from the outcome of this
uncertainty.
Basis for Opinion
These consolidated financial statements are the
responsibility of the Company’s management. Our responsibility is to express an opinion on the consolidated financial statements
based on our audit. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB)
and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable
rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the
standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated
financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we
engaged to perform, an audit of its internal control over financial reporting. As part of our audit, we are required to obtain an understanding
of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s
internal control over financial reporting. Accordingly, we express no such opinion.
Our audit included performing procedures to assess
the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures
that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the
consolidated financial statements. Our audit also included evaluating the accounting principles used and significant estimates made by
management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audit provides
a reasonable basis for our opinion.
/s/ Haskell & White LLP
We have served as the Company’s auditor since 2023.
Irvine, California
March 29, 2024
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING
FIRM
To the Board of Directors and
Stockholders of Tenon Medical, Inc. and Subsidiary
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheet of Tenon
Medical, Inc and Subsidiary (collectively the “Company”) as of December 31, 2022, and the related consolidated statements
of operations and comprehensive loss, consolidated statements of convertible preferred stock and stockholders’ equity (deficit),
and consolidated statements of cash flows for the year then ended, and the related notes (collectively referred to as the consolidated
financial statements).
In our opinion, the consolidated financial statements present fairly,
in all material respects, the financial position of the Company as of December 31, 2022, and the results of its operations and its cash
flows for the year ended December 31, 2022, in conformity with accounting principles generally accepted in the United States of America.
Going Concern
The 2022 consolidated financial statements were prepared assuming that
the Company would continue as a going concern. As of December 31, 2022, the Company had suffered recurring losses from operations, incurred
negative cash flows from operating activities, and had stated that substantial doubt exists about the Company’s ability to continue
as a going concern. The 2022 consolidated financial statements did not include any adjustments that might result from the outcome of this
uncertainty.
Basis for Opinion
These consolidated financial statements are the responsibility of the
Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based
on our audit. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and
are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules
and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB.
Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements
are free of material misstatement, whether due to error or fraud.
Our audit of the consolidated financial statements included performing
procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing
procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures
in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates
made by management, as well as evaluating the overall presentation of the consolidated financial statements. Our audit also included performing
such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
/s/ Armanino LLP
San Jose, California
March 10, 2023
We began serving as the Company’s auditor
in 2021. In 2023, we became the predecessor auditor.
Tenon Medical, Inc.
Consolidated Balance Sheets
(In thousands, except share data)
| | |
December 31, | | |
December 31, | |
| | |
2023 | | |
2022 | |
| Assets | |
| | |
| |
| Current assets: | |
| | |
| |
| Cash and cash equivalents | |
$ | 2,428 | | |
$ | 2,129 | |
| Short-term investments | |
| - | | |
| 6,441 | |
| Accounts receivable | |
| 518 | | |
| 228 | |
| Inventory, net | |
| 554 | | |
| 415 | |
| Prepaid expenses | |
| 389 | | |
| 134 | |
| Total current assets | |
| 3,889 | | |
| 9,347 | |
| Fixed assets, net | |
| 961 | | |
| 793 | |
| Deposits | |
| 51 | | |
| 51 | |
| Operating lease right-of-use asset | |
| 646 | | |
| 873 | |
| Deferred offering costs | |
| 798 | | |
| 25 | |
| TOTAL ASSETS | |
$ | 6,345 | | |
$ | 11,089 | |
| | |
| | | |
| | |
| Liabilities and Stockholders’ EQUITY | |
| | | |
| | |
| Current liabilities: | |
| | | |
| | |
| Accounts payable | |
$ | 433 | | |
$ | 550 | |
| Accrued expenses | |
| 808 | | |
| 717 | |
| Current portion of accrued commissions | |
| 470 | | |
| 1,035 | |
| Current portion of operating lease liability | |
| 256 | | |
| 228 | |
| Convertible notes payable and accrued interest, net of debt discount of $77 and $0 at December 31, 2023 and 2022, respectively | |
| 1,173 | | |
| - | |
| Total current liabilities | |
| 3,140 | | |
| 2,530 | |
| Accrued commissions, net of current portion | |
| 1,999 | | |
| 1,624 | |
| Operating lease liability, net of current portion | |
| 428 | | |
| 683 | |
| Total liabilities | |
| 5,567 | | |
| 4,837 | |
| | |
| | | |
| | |
| Commitments and contingencies (Notes 6 and 10) | |
| | | |
| | |
| Stockholders’ equity: | |
| | | |
| | |
| Common stock, $0.001 par value; 130,000,000 shares authorized at December 31, 2023 and 2022; 2,600,311 and 1,123,680 shares issued and outstanding at December 31, 2023 and 2022, respectively | |
| 3 | | |
| 1 | |
| Additional paid-in capital | |
| 55,894 | | |
| 45,843 | |
| Accumulated deficit | |
| (55,073 | ) | |
| (39,492 | ) |
| Accumulated other comprehensive loss | |
| (46 | ) | |
| (100 | ) |
| Total stockholders’ equity | |
| 778 | | |
| 6,252 | |
| TOTAL LIABILITIES AND STOCKHOLDERS’ EQUITY | |
$ | 6,345 | | |
$ | 11,089 | |
The accompanying notes are an integral part
of these consolidated financial statements.
See Reports of Independent Registered Public
Accounting Firms.
Tenon Medical, Inc.
Consolidated Statements of Operations and Comprehensive
Loss
(In thousands, except per share data)
| | |
Years Ended December 31, | |
| | |
2023 | | |
2022 | |
| Revenue | |
$ | 2,928 | | |
$ | 691 | |
| Cost of sales | |
| 1,687 | | |
| 1,332 | |
| Gross Profit (Loss) | |
| 1,241 | | |
| (641 | ) |
| | |
| | | |
| | |
| Operating Expenses | |
| | | |
| | |
| Research and development | |
| 3,163 | | |
| 2,828 | |
| Sales and marketing | |
| 6,778 | | |
| 7,833 | |
| General and administrative | |
| 7,027 | | |
| 7,423 | |
| Total Operating Expenses | |
| 16,968 | | |
| 18,084 | |
| | |
| | | |
| | |
| Loss from Operations | |
| (15,727 | ) | |
| (18,725 | ) |
| | |
| | | |
| | |
| Other Income (Expense) | |
| | | |
| | |
| Gain on investments | |
| 167 | | |
| 180 | |
| Interest expense | |
| (21 | ) | |
| (354 | ) |
| Other expense, net | |
| - | | |
| (18 | ) |
| Total Other Income (Expense), net | |
| 146 | | |
| (192 | ) |
| Net Loss | |
$ | (15,581 | ) | |
$ | (18,917 | ) |
| Net Loss Per Share of Common Stock | |
| | | |
| | |
Basic and diluted | |
$ | (8.59 | ) | |
$ | (23.62 | ) |
| | |
| | | |
| | |
| Weighted-Average Shares of Common Stock Outstanding | |
| | | |
| | |
Basic and diluted | |
| 1,814 | | |
| 801 | |
| | |
| | | |
| | |
| Consolidated Statements of Comprehensive Loss: | |
| | | |
| | |
| Net loss | |
$ | (15,581 | ) | |
$ | (18,917 | ) |
| Unrealized loss on investments | |
| 16 | | |
| (16 | ) |
| Foreign currency translation adjustment | |
| 38 | | |
| 7 | |
| Total Comprehensive Loss | |
$ | (15,527 | ) | |
$ | (18,926 | ) |
The accompanying notes are an integral part
of these consolidated financial statements.
See Reports of Independent Registered Public
Accounting Firms.
Tenon Medical, Inc.
Consolidated Statements of Convertible Preferred
Stock and Stockholders’ Equity (Deficit)
(In thousands, except share data)
| | |
Series
A
Convertible
Preferred Stock | | |
Series
B
Convertible
Preferred Stock | | |
Common
Stock | | |
Additional
Paid-In | | |
Accumulated | | |
Accumulated
Other
Comprehensive | | |
| |
| | |
Shares | | |
Amount | | |
Shares | | |
Amount | | |
Shares | | |
Amount | | |
Capital | | |
Deficit | | |
Loss | | |
Total | |
| Balance at December
31, 2021 | |
| 2,550,763 | | |
$ | 12,367 | | |
| 491,222 | | |
$ | 1,272 | | |
| 98,995 | | |
$ | - | | |
$ | 114 | | |
$ | (20,575 | ) | |
$ | (91 | ) | |
$ | (20,552 | ) |
| Stock-based compensation expense | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| 2,897 | | |
| - | | |
| - | | |
| 2,897 | |
| Issuance of common stock and warrants,
net of issuance costs | |
| - | | |
| - | | |
| - | | |
| - | | |
| 320,000 | | |
| - | | |
| 13,765 | | |
| - | | |
| - | | |
| 13,765 | |
| Common stock issued upon conversion
of Series A preferred stock | |
| (2,550,763 | ) | |
| (12,367 | ) | |
| - | | |
| - | | |
| 244,773 | | |
| - | | |
| 12,367 | | |
| - | | |
| - | | |
| 12,367 | |
| Common stock issued upon conversion
of Series B preferred stock | |
| - | | |
| - | | |
| (491,222 | ) | |
| (1,272 | ) | |
| 24,561 | | |
| - | | |
| 1,272 | | |
| - | | |
| - | | |
| 1,272 | |
| Common stock issued upon conversion
of debt | |
| - | | |
| - | | |
| - | | |
| - | | |
| 395,542 | | |
| 1 | | |
| 13,867 | | |
| - | | |
| - | | |
| 13,868 | |
| Common stock issued for services | |
| - | | |
| - | | |
| - | | |
| - | | |
| 39,809 | | |
| - | | |
| 1,561 | | |
| - | | |
| - | | |
| 1,561 | |
| Other comprehensive loss | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| (9 | ) | |
| (9 | ) |
| Net loss | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| (18,917 | ) | |
| - | | |
| (18,917 | ) |
| Balance at December 31, 2022 | |
| - | | |
$ | - | | |
| - | | |
$ | - | | |
| 1,123,680 | | |
$ | 1 | | |
$ | 45,843 | | |
$ | (39,492 | ) | |
$ | (100 | ) | |
$ | 6,252 | |
| Stock-based compensation expense | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| 4,145 | | |
| | | |
| | | |
| 4,145 | |
| Release of restricted stock units | |
| - | | |
| - | | |
| - | | |
| - | | |
| 61,200 | | |
| - | | |
| - | | |
| | | |
| | | |
| - | |
| Issuance of common stock and warrants,
net of issuance costs | |
| - | | |
| - | | |
| - | | |
| - | | |
| 1,000,000 | | |
| 1 | | |
| 4,807 | | |
| | | |
| | | |
| 4,808 | |
| Issuance of common stock, net of
issuance costs | |
| - | | |
| - | | |
| - | | |
| - | | |
| 232,100 | | |
| 1 | | |
| 494 | | |
| | | |
| | | |
| 495 | |
| Common stock issued for services | |
| - | | |
| - | | |
| - | | |
| - | | |
| 98,909 | | |
| - | | |
| 289 | | |
| | | |
| | | |
| 289 | |
| Issuance of common stock upon exercise
of warrants | |
| - | | |
| - | | |
| - | | |
| - | | |
| 82,000 | | |
| - | | |
| 258 | | |
| | | |
| | | |
| 258 | |
| Warrants issued in connection with
convertible debt | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| 58 | | |
| | | |
| | | |
| 58 | |
| Shares issued for reverse stock split | |
| - | | |
| - | | |
| - | | |
| - | | |
| 2,422 | | |
| - | | |
| - | | |
| | | |
| | | |
| - | |
| Other comprehensive income | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| | | |
| 54 | | |
| 54 | |
| Net loss | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| (15,581 | ) | |
| | | |
| (15,581 | ) |
| Balance at
December 31, 2023 | |
| - | | |
$ | - | | |
| - | | |
$ | - | | |
| 2,600,311 | | |
$ | 3 | | |
$ | 55,894 | | |
$ | (55,073 | ) | |
$ | (46 | ) | |
$ | 778 | |
The accompanying notes are an integral part
of these consolidated financial statements.
See Reports of Independent Registered Public
Accounting Firms.
Tenon Medical, Inc.
Consolidated Statements of Cash Flows
(In thousands)
| | |
Years Ended
December 31, | |
| | |
2023 | | |
2022 | |
| Cash Flows from Operating Activities | |
| | |
| |
| Net loss | |
$ | (15,581 | ) | |
$ | (18,917 | ) |
| Adjustments to reconcile net loss to net cash used in operating activities: | |
| | | |
| | |
| Unrealized loss on investments | |
| - | | |
| (16 | ) |
| Non-cash interest expense | |
| - | | |
| 362 | |
| Stock-based compensation expense | |
| 4,145 | | |
| 2,897 | |
| Common stock issued for services | |
| - | | |
| 1,561 | |
| Depreciation and amortization | |
| 199 | | |
| 78 | |
| Loss on write-off of fixed assets | |
| - | | |
| 77 | |
| Amortization of operating right-of-use asset | |
| 227 | | |
| 211 | |
| Increase (decrease) in cash resulting from changes in: | |
| | | |
| | |
| Accounts receivable | |
| (290 | ) | |
| (152 | ) |
| Inventory | |
| (139 | ) | |
| (227 | ) |
| Prepaid expenses and other assets | |
| (301 | ) | |
| (57 | ) |
| Accounts payable | |
| (117 | ) | |
| 72 | |
| Accrued expenses | |
| (99 | ) | |
| 2,288 | |
| Operating lease liability | |
| (227 | ) | |
| (202 | ) |
| Net cash used in operating activities | |
| (12,183 | ) | |
| (12,025 | ) |
| | |
| | | |
| | |
| Cash Flows from Investing Activities | |
| | | |
| | |
| Sales of short-term investments | |
| 6,996 | | |
| 8,079 | |
| Purchases of short-term investments | |
| (493 | ) | |
| (10,116 | ) |
| Purchases of property and equipment | |
| (361 | ) | |
| (847 | ) |
| Net cash provided by (used in) investing activities | |
| 6,142 | | |
| (2,884 | ) |
| | |
| | | |
| | |
| Cash Flows from Financing Activities | |
| | | |
| | |
| Proceeds from issuance of common stock and warrants, net of issuance costs | |
| 4,808 | | |
| 14,139 | |
| Proceeds from issuance of common stock, net of issuance costs | |
| 495 | | |
| - | |
| Proceeds from issuance of convertible notes payable | |
| 1,250 | | |
| - | |
| Proceeds from exercise of warrants | |
| 258 | | |
| - | |
| Deferred offering costs | |
| (509 | ) | |
| (25 | ) |
| Net cash provided by financing activities | |
| 6,302 | | |
| 14,114 | |
| | |
| | | |
| | |
| Effect of foreign currency translation on cash flow | |
| 38 | | |
| 7 | |
| Net Increase (Decrease) in Cash and Cash Equivalents | |
| 299 | | |
| (788 | ) |
| | |
| | | |
| | |
| Cash and Cash Equivalents at Beginning of Year | |
| 2,129 | | |
| 2,917 | |
| Cash and Cash Equivalents at End of Year | |
$ | 2,428 | | |
$ | 2,129 | |
| | |
| | | |
| | |
| Cash at End of Year | |
$ | 2,428 | | |
$ | 480 | |
| Cash Equivalents at End of Year | |
$ | - | | |
$ | 1,649 | |
| | |
| | | |
| | |
| Supplemental Disclosures of Cash Flow Information | |
| | | |
| | |
| Cash paid during the year for: | |
| | | |
| | |
| Interest | |
$ | - | | |
$ | - | |
| Income taxes | |
$ | - | | |
$ | - | |
| | |
| | | |
| | |
| Non-cash investment and financing activities: | |
| | | |
| | |
| Common stock issued upon conversion of preferred stock | |
$ | - | | |
$ | 13,639 | |
| Common stock issued upon conversion of debt | |
$ | - | | |
$ | 13,868 | |
The accompanying notes are an integral part
of these consolidated financial statements.
See Reports of Independent Registered Public
Accounting Firms.
Notes to Consolidated Financial Statements (in
thousands, except share and per-share data)
1. Organization and Business
Nature of operations
Tenon Medical, Inc. (the “Company”)
was incorporated in the State of Delaware on June 19, 2012 and was headquartered in San Ramon, California until June 2021 when it relocated
to Los Gatos, California. The Company is a medical device company that has developed The Catamaran™ SI Joint Fusion System (“the
Catamaran System”) that offers a novel, less invasive approach to the sacroiliac joint (the “SI Joint”) using a single,
robust, titanium implant for treatment of the most common types of SI Joint disorders that cause lower back pain. The Company received
U.S. Food and Drug Administration (“FDA”) clearance in 2018 for The Catamaran System and is currently focused on the U.S.
market. Since the national launch of the Catamaran System in October 2022, the Company is focused on three commercial opportunities: 1)
Primary SI Joint procedures, 2) Revision procedures of failed SI Joint implants and 3) SI Joint fusion adjunct to a spine fusion construct.
Basis of consolidation
The condensed financial statements of the Company
include the accounts of the Company and its wholly-owned subsidiary, Tenon Technology AG (“TTAG”), a Swiss company. All intercompany
balances and transactions have been eliminated in consolidation. The financial statements of TTAG are prepared for the same reporting
period as the parent, using consistent accounting policies in all material respects.
2. Summary of Significant Accounting Principles
Basis of presentation
The accompanying consolidated financial statements
have been prepared on the accrual basis in accordance with generally accepted accounting principles as promulgated in the United States
of America (“U.S. GAAP”).
Going concern uncertainty and liquidity requirements
The accompanying consolidated financial statements
have been prepared assuming the Company will continue as a going concern, which contemplates the realization of assets and the settlement
of liabilities and commitments in the normal course of business. There is substantial doubt about the Company’s ability to continue
as a going concern for one year after the date that these financial statements are issued.
Since inception, the Company has incurred losses
and negative cash flows from operations. Management expects to incur additional operating losses and negative cash flows from operations
in the foreseeable future as the Company continues its product development programs and the commercialization of The Catamaran System.
Based on the Company’s expected level of revenues and expenditures, the Company believes that its existing cash and cash equivalents
as of December 31, 2023 will not provide sufficient funds to enable it to meet its obligations for a period of at least twelve months
from the date of the filing of these consolidated financial statements. The Company plans to raise the necessary additional capital through
one or a combination of public or private equity offerings, debt financings, and collaborations (see Note 13). The consolidated financial
statements do not include any adjustments that might result from the outcome of this uncertainty.
Use of estimates
The preparation of the consolidated financial
statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect certain reported amounts and
disclosures. Accordingly, actual results could differ from those estimates. Significant estimates made by management include, but are
not limited to, realization of deferred tax assets, accrued liabilities, obsolescence of inventory, the fair value of accrued commissions
and stock-based compensation.
Reverse Stock Splits
On April 6, 2022, the Company effected a 1-for-2
reverse stock split (the “2022 Reverse Stock Split”) by filing an amendment to the Company’s Amended and Restated Certificate
of Incorporation, as amended, with the Delaware Secretary of State. The 2022 Reverse Stock Split combined every two shares of our common
stock issued and outstanding immediately prior to effecting the 2022 Reverse Stock Split into one share of common stock. Similarly, shares
of Series A and Series B Preferred Stock became convertible into common stock at a conversion rate of one-to-0.5, subject to adjustments
for stock dividends, splits, combinations, and similar events. No fractional shares were issued in connection with the 2022 Reverse Stock
Split.
On November 2, 2023, the Company effected a 1-for-10
reverse stock split (the “2023 Reverse Stock Split”) by filing an amendment to the Company’s Amended and Restated Certificate
of Incorporation, as amended, with the Delaware Secretary of State. The 2023 Reverse Stock Split combined every ten shares of our common
stock issued and outstanding immediately prior to effecting the 2023 Reverse Stock Split into one share of common stock. No fractional
shares were issued in connection with the 2023 Reverse Stock Split. All historical share and per share amounts reflected throughout this
document have been adjusted to reflect the 2022 Reverse Stock Split and the 2023 Reverse Stock Split. The authorized number of shares
and the par value per share of the Company’s common stock were not affected by the 2022 Reverse Stock Split or the 2023 Reverse
Stock Split.
Segments
The Company operates in one business segment.
Although the Company’s Swiss subsidiary is located in a different geographical area, management uses one measurement of profitability
and does not segregate its business for internal reporting.
Cash and cash equivalents
The Company considers all highly liquid investments
with maturities of 90 days or less at the date of purchase to be cash equivalents.
Investments
The Company classifies its investments in marketable
securities as available-for-sale and records them at fair value in its consolidated balance sheets. The net unrealized gains and losses
are recorded as a separate component of stockholders’ equity. Realized gains and losses are recorded in the consolidated statements
of operations and comprehensive loss. The Company determines any realized gains or losses on the sale of marketable debt securities on
a specific identification method and records such gains and losses as a component of other income (expense) net.
Accounts receivable and allowance for doubtful
accounts
Accounts receivable are derived from products
delivered to customers and are stated at their net realizable value. The Company records an allowance for estimated uncollectible accounts
in an amount approximating anticipated losses. Individual uncollectible accounts are written off against the allowance when collection
of the individual accounts appears doubtful. In determining the amount of the allowance, the Company considers its historical level of
credit losses. The Company also makes judgments about the creditworthiness of significant customers based on ongoing credit evaluations,
and the Company assesses current economic trends that might impact the level of credit losses in the future. Historically, the Company
has had no significant write-offs of accounts receivable. However, since the Company cannot reliably predict future changes in the financial
stability of its customers, it cannot guarantee that its allowances will continue to be adequate. If actual credit losses are significantly
greater than the allowance, the Company would increase its general and administrative expenses and increase its reported net losses. As
of December 31, 2023 and 2022, the Company’s allowance for expected credit losses was $0.
Inventory
Inventory is stated at lower of cost or net realizable
value. The Company establishes the inventory basis by determining the cost based on standard costs approximating the purchase costs on
a first-in, first-out basis. The excess and obsolete inventory is estimated based on future demand and market conditions. Inventory write-downs
are charged to cost of goods sold. As of December 31, 2023 and 2022, inventory consisted of finished goods and raw materials.
Deferred offering costs
Deferred offering costs, which consist of direct
incremental legal, consulting, banking, and accounting fees relating to the Company’s future offerings, are capitalized, and are
offset against proceeds received upon the effectiveness of the offering. In the event an anticipated offering is terminated, deferred
offering costs will be expensed.
Fixed assets, net
Fixed assets are stated at cost less accumulated
depreciation. Depreciation is computed using the straight-line method over the estimated useful lives of the assets. Equipment, computers,
software, and furniture and fixtures are depreciated over periods ranging from three to seven years, and leasehold improvements over the
shorter of the lease term or the life of the asset. Construction in progress pertains to the cost of individual components of a custom
instrument set used for surgical placement of the Company’s products that have not yet been placed into service. The cost of maintenance
and repairs is charged to expense as incurred; significant renewals and betterments are capitalized. Deductions are made for retirements
resulting from renewals or betterments.
Leases
The Company leases its headquarters in Los Gatos,
California. At the inception of a contract, the Company assesses whether that contract is, or contains, a lease. The Company’s assessment
is based on: (1) whether the contract involves the use of a distinct identified asset, (2) whether the Company obtains the right to substantially
all the economic benefit from the use of the asset throughout the term, and (3) whether the Company has the right to direct the use of
the asset. At inception of a lease, the Company allocates the consideration in the contract to each lease and non-lease component based
on the component’s relative stand-alone price to determine the lease payments. Lease and non-lease components are accounted for
separately.
Leases are classified as either finance leases
or operating leases based on criteria in FASB ASC 842, “Leases”. The Company’s facility lease is classified as an operating
lease. Right-of-use assets represent the Company’s right to use an underlying asset for the lease term and lease liabilities represent
the obligation to make lease payments arising from the lease. Operating lease right-of-use assets and liabilities are recognized at the
lease’s commencement date based on the present value of lease payments over the lease term. When a lease did not provide an implicit
rate, the Company used its estimated incremental borrowing rate based on the information available at the commencement date in determining
the present value of future payments. The Company has elected not to recognize ROU assets and lease liabilities for short-term operating
leases that have a term of 12 months or less. Lease expense for operating leases is recognized on a straight-line basis over the lease
term and is included in operating expenses in the consolidated statements of operations and comprehensive loss.
Long-lived assets
The Company regularly reviews the carrying value
and estimated lives of all of its long-lived assets, including property and equipment, to determine whether indicators of impairment may
exist that warrant adjustments to carrying values or estimated useful lives. The determinants used for this evaluation include management’s
estimate of the asset’s ability to generate positive income from operations and positive undiscounted cash flow in future periods
as well as the strategic significance of the assets to the Company’s business objectives.
Fair value measurements
In accordance with Accounting Standards Codification
(“ASC”) 820, Fair Value Measurement, fair value is the price that would be received from selling an asset or paid to transfer
a liability (i.e., the exit price) in an orderly transaction between market participants at the measurement date. ASC 820 establishes
a fair value hierarchy for inputs used in measuring fair value that maximizes the use of observable inputs and minimizes the use of unobservable
inputs by requiring that the most observable inputs be used when available.
Observable inputs are those that market participants
would use in pricing the asset or liability based on market data obtained from sources independent of the Company. Unobservable inputs
reflect the Company’s assumptions about the inputs that market participants would use in pricing the asset or liability based on
the best information available in the circumstances.
The fair value hierarchy is categorized into three
levels based on the inputs as follows:
Level 1 – Quoted
prices are available in active markets for identical assets or liabilities as of the reported date.
Level 2 – Pricing
inputs are other than quoted prices in active markets, which are either directly or indirectly observable as of the reported date. The
nature of these financial instruments includes cash instruments for which quoted prices are available but are traded less frequently,
derivative instruments whose fair values have been derived using a model where inputs to the model are directly observable in the market
and instruments that are fair valued using other financial instruments, the parameters of which can be directly observed.
Level 3 – Instruments
that have little to no pricing observability as of the measurement date. These financial instruments are measured using management’s
best estimate of fair value, where the inputs into the determination of fair value require significant management judgment or estimation.
The degree of judgment exercised by the Company
in determining fair value is greatest for assets categorized in Level 3. In certain cases, the inputs used to measure fair value may fall
into different levels of the fair value hierarchy. In such cases, for disclosure purposes, the level in the fair value hierarchy within
which the fair value measurement falls in its entirety is determined by the lowest level input that is significant to the fair value measurement.
Income taxes
Income taxes are recorded in accordance with Financial
Accounting Standards Board (“FASB”) ASC Topic 740, Income Taxes (“ASC 740”), which provides for deferred taxes
using an asset and liability approach. Under this method, the Company records deferred tax assets and liabilities for the expected future
tax consequences of temporary differences between the financial statement carrying amounts and the tax basis of assets and liabilities
using enacted tax rates expected to be in effect when the differences are expected to reverse. Valuation allowances are provided when
necessary to reduce net deferred tax assets to the amount that is more likely than not to be realized. Based on the available evidence,
the Company is unable, at this time, to support the determination that it is more likely than not that its deferred tax assets will be
utilized in the future. Accordingly, the Company recorded a full valuation allowance as of December 31, 2023 and 2022. The Company intends
to maintain valuation allowances until sufficient evidence exists to support its reversal.
Current income taxes are based upon the year’s
income taxable for federal, state, and foreign tax reporting purposes. Deferred income taxes are provided for certain income and expenses,
which are recognized in different periods for tax and financial reporting purposes.
The Company’s policy is not to record deferred
income taxes on the undistributed earnings of foreign subsidiaries that are indefinitely reinvested in foreign operations.
Revenue recognition
The Company’s revenue is derived from the
sale of its products to medical groups and hospitals in the United States. Revenue is recognized when control is transferred to the customer,
in an amount that reflects the consideration we expect to be entitled to in exchange for the goods or services, using the following five
step approach: (1) identify the contract with a customer, (2) identify the performance obligations in the contract, (3) determine the
transaction price, (4) allocate the transaction price to the performance obligations in the contract, and (5) recognize revenue when a
performance obligation is satisfied.
The Company generates revenue from the sale of
products to hospitals or medical facilities where its products are delivered in advance of a procedure. The performance obligation is
the delivery of the products along with the completion of the surgery and therefore, revenue is recognized upon delivery to the customers
and completion of the surgery, net of rebates and price discounts. The Company accounts for rebates and price discounts as a reduction
to revenue, calculated based on the terms agreed to with the customer. Historically, there have been no significant rebates or price discounts.
Sales prices are specified prior to the transfer of control to the customer, via either the customer contract, agreed price list, purchase
order, or written communication with the customer. Prior to October 2022, the Company had an agreement in place with a national distributor,
which included standard terms that did not allow for payment contingent on resale of the product, obtaining financing, or other terms
that could impact the distributor’s payment obligation. The Company billed and collected directly with the end-user customers and
recognized revenue based on the gross sales price. For direct sales to end-user customers, the Company’s standard payment terms
are generally net 30 days.
The Company offers its standard warranty to all
customers and does not sell any warranties on a standalone basis. The Company’s warranty provides that its products are free of
material defects and conform to specifications, and includes an offer to replace or refund the purchase price of defective products. This
assurance does not constitute a service and is not considered a separate performance obligation. The Company estimates warranty liabilities
at the time of revenue recognition and records them as a charge to cost of goods sold.
Contract modifications generally do not occur
during the performance of the Company’s contracts.
Payments received prior to satisfying the revenue
recognition criteria are recorded as deferred revenue on the consolidated balance sheets. As of December 31, 2023 and 2022, there were
no remaining performance obligations that would give rise to deferred revenue.
Sales commissions are recorded in sales and marketing
expenses during the same period as the corresponding revenues.
Research and development
The Company engages in improving existing products
and new product development efforts. Research and development expenses relating to these efforts are expensed as incurred.
Stock-based compensation
The Company accounts for all stock-based compensation
awards using a fair-value method on the grant date and recognizes the fair value of each award as an expense over the requisite service
period.
The Company recognizes compensation costs related
to stock-based awards granted to employees, directors, and consultants including stock options, based on the estimated fair value of the
awards on the date of grant. We estimate the grant date fair value, and the resulting stock-based compensation, using the Black-Scholes
option-pricing model. The grant date fair value of the stock-based awards is generally recognized on a straight-line basis over the requisite
service period, which is generally the vesting period of the respective awards.
The Black-Scholes option-pricing model requires
the use of subjective assumptions to determine the fair value of stock-based awards. These assumptions include:
Expected Term-The expected term represents
the period that stock-based awards are expected to be outstanding. The expected term for option grants is determined using the simplified
method. The simplified method deems the expected term to be the midpoint between the vesting date and the contractual life of the stock-based
awards.
Expected Volatility-Since the Company
has only been publicly held since April 2022 and does not have any trading history for its common stock prior to that date, the expected
volatility was estimated based on the average volatility for comparable publicly traded companies over a period equal to the expected
term of the stock option grants. The comparable companies were chosen based on their similar size, stage in the life cycle, or area of
specialty.
Risk-Free Interest Rate-The risk-free
interest rate is based on the U.S. Treasury zero coupon issues in effect at the time of grant for periods corresponding with the expected
term of option.
Expected Dividends-The Company has
never paid dividends on its common stock and has no plans to pay dividends on its common stock. Therefore, an expected dividend yield
of zero is used.
The Company account for forfeitures as they occur.
The Company’s board of directors intends
all options granted to be exercisable at a price per share not less than the per share fair value of our common stock underlying those
options on the date of grant.
Prior to the Company’s initial public offering,
the estimated fair value of its common stock was determined at each valuation date by a third-party independent valuation firm in accordance
with the guidelines outlined in the American Institute of Certified Public Accountants Practice Aid, Valuation of Privately-Held-Company
Equity Securities Issued as Compensation. These valuations took into account numerous factors, including developments at our company and
market conditions.
The May 21, 2021 valuation used a hybrid method
which combines the Probability Weighted Expected Return Method (“PWERM”) with the OPM. The PWERM considers a set of discrete
potential liquidity scenarios for the Company, the value common stock would receive in each scenario, and the time required and risk inherent
in achieving those values. The May 21, 2021 valuation examined the following scenarios for the Company: (i) an IPO; (ii) remaining private
and raising capital; and (iii) dissolution. Within the IPO scenario, 100% weighting was placed on the Market Approach for determining
the enterprise value. The Market Approach assumes that businesses operating in the same industry will share similar characteristics, and
therefore a comparison of the business to similar businesses whose financial information is publicly available may provide a reasonable
basis to estimate a subject business’s value. The equity value in the IPO scenario was estimated considering guideline IPOs, the
anticipated size of the Company’s offering, and forecasted cash and debt. The estimated common stock value as of the IPO was present
valued using a discount rate of 22.4% based on Company’s WACC, less an adjustment of 2.0% to reflect the risk reduction of an IPO
event.
The August 31, 2021 valuation used a hybrid method
which combines the Probability Weighted Expected Return Method (“PWERM”) with the OPM. The PWERM considers a set of discrete
potential liquidity scenarios for the Company, the value common stock would receive in each scenario, and the time required and risk inherent
in achieving those values. The August 31, 2021 valuation examined the following scenarios for the Company: (i) an IPO; (ii) remaining
private and raising capital; and (iii) dissolution. Within the IPO scenario, 100% weighting was placed on the Market Approach for determining
the enterprise value. The Market Approach assumes that businesses operating in the same industry will share similar characteristics, and
therefore a comparison of the business to similar businesses whose financial information is publicly available may provide a reasonable
basis to estimate a subject business’s value. The equity value in the IPO scenario was estimated considering guideline IPOs, the
anticipated size of the Company’s offering, and forecasted cash and debt. The estimated common stock value as of the IPO was present
valued using a discount rate of 32.0% based on Company’s WACC, less an adjustment of 5.0% to reflect the risk reduction of an IPO
event.
The October 28, 2021 valuation used a hybrid method
which combines the Probability Weighted Expected Return Method (“PWERM”) with the OPM. The PWERM considers a set of discrete
potential liquidity scenarios for the Company, the value common stock would receive in each scenario, and the time required and risk inherent
in achieving those values. The October 28, 2021 valuation examined the following scenarios for the Company: (i) an IPO; (ii) remaining
private and raising capital; and (iii) dissolution. Within the IPO scenario, 100% weighting was placed on the Market Approach for determining
the enterprise value. The Market Approach assumes that businesses operating in the same industry will share similar characteristics, and
therefore a comparison of the business to similar businesses whose financial information is publicly available may provide a reasonable
basis to estimate a subject business’s value. The equity value in the IPO scenario was estimated considering guideline IPOs, the
anticipated size of the Company’s offering, and forecasted cash and debt. The estimated common stock value as of the IPO was present
valued using a discount rate of 27.2% based on Company’s WACC, less an adjustment of 5.0% to reflect the risk reduction of an IPO
event.
In determining the enterprise value within the
remain private scenario, 100% weighting was applied to the DCF Method under the income approach, in the same manner as in the December
31, 2018, 2019, and 2020 valuations. The discount rate in this scenario was determined to be 22.4% based on Company’s WACC. Adjustments
were made to the enterprise value for the Company’s cash and debt as of the valuation date to determine the equity value in this
scenario. The OPM was used to allocate the equity value to our common stock. The equity volatility rate was determined to be 70.0% based
on the volatility rate of certain comparable public companies. DLOMs of (i) 10.0% in the IPO scenario and (ii) 30.0% in the remaining
private scenario were applied to the common stock.
Following the closing of the initial public offering,
the fair value of the Company’s common stock was determined based on the closing price of its common stock on the Nasdaq Capital
Market.
Foreign currency translation and other comprehensive
income
The functional currency of Tenon Technology AG
is the Swiss franc. Accordingly, TTAG’s assets and liabilities are translated from their respective functional currency into U.S.
Dollars at period-end rates, and TTAG’s revenue and expenses are translated at the weighted-average exchange rate for the period.
Adjustments resulting from this translation process are classified as other comprehensive income or loss and shown as a separate component
of equity.
When intercompany foreign currency transactions
between entities included in the consolidated financial statements are of a long-term investment nature (i.e., those for which settlement
is not planned or anticipated in the foreseeable future) foreign currency translation adjustments resulting from those transactions are
included in stockholders’ equity (deficit) as accumulated other comprehensive loss or income. When intercompany transactions are
deemed to be of a short-term nature, translation adjustments are required to be included in the consolidated statements of operations.
Net loss per share
Basic net loss per share is based upon the weighted-average
number of common shares outstanding. Diluted net loss per share is based on the assumption that all potential common stock equivalents
(convertible preferred stock, stock options, and warrants) are converted or exercised. The calculation of diluted net loss per share excludes
potential common stock equivalents if the effect is anti-dilutive. For the periods presented, the Company’s weighted-average common
shares outstanding for basic and diluted are the same because the effect of the potential common stock equivalents is anti-dilutive.
The Company had the following dilutive common
stock equivalents as of December 31, 2023 and 2022 which were excluded from the calculation because their effect was anti-dilutive.
| | |
December 31,
2023 | | |
December 31,
2022 | |
| Outstanding restricted stock units | |
| 76,916 | | |
| 131,858 | |
| Outstanding stock options | |
| 102,089 | | |
| 89,889 | |
| Outstanding warrants | |
| 1,927,600 | | |
| 9,600 | |
| Total | |
| 2,106,605 | | |
| 231,347 | |
Adoption of New Accounting Pronouncements
In June 2016, the Financial Accounting Standards
Board issued Accounting Standards Update 2016-13, “Financial Instruments-Credit Losses (Topic 326): Measurement of Credit
Losses on Financial Instruments”. This standard requires an impairment model (known as the current expected credit loss (“CECL”)
model) that is based on expected losses rather than incurred losses. Under the new guidance, each reporting entity should estimate an
allowance for expected credit losses, which is intended to result in more timely recognition of losses. The Company adopted this guidance
effective January 1, 2023. The adoption of this guidance did not have a significant impact on the Company’s consolidated financial
statements or results of operations.
3. Investments
The following table sets forth by level, within
the fair value hierarchy, the Company’s investments at fair value as of December 31, 2023 and 2022:
| | |
Level 2 | |
| Corporate debt securities: | |
| |
| December 31, 2023 | |
$ | - | |
| December 31, 2022 | |
$ | 6,441 | |
Cost and fair value of available-for-sale investments
as of December 31, 2023 and 2022 are as follows:
| | |
Amortized
Cost | | |
Gross
Unrealized
Gains | | |
Gross Unrealized Losses | | |
Fair
Value | |
| Corporate debt securities: | |
| | |
| | |
| | |
| |
| December 31, 2023 | |
$ | - | | |
$ | - | | |
$ | - | | |
$ | - | |
| December 31, 2022 | |
$ | 6,457 | | |
$ | - | | |
$ | (16 | ) | |
$ | 6,441 | |
All of the investments with gross unrealized losses
have been in a continuous loss position for less than 12 months.
During the years ended December 31, 2023 and 2022,
the Company did not recognize any significant other-than-temporary impairment losses because the Company does not intend to sell the investments
before recovery of their amortized cost bases.
During the years ended December 31, 2023 and 2022,
there were net gains of approximately $167 and $180, respectively, included in the Company’s net loss. Accrued interest as of December
31, 2023 and 2022 was approximately $8 and $13, respectively, and is included in prepaid expenses in the Company’s consolidated
balance sheets.
4. Inventory, net
Inventory, net of reserves, consisted of the following:
| | |
December 31,
2023 | | |
December 31,
2022 | |
| Raw materials | |
$ | 22 | | |
$ | 9 | |
| Finished goods | |
| 532 | | |
| 406 | |
| Inventory | |
$ | 554 | | |
$ | 415 | |
5. Fixed Assets, net
Fixed assets, net, consisted of the following:
| | |
December 31,
2023 | | |
December 31,
2022 | |
| Construction in progress | |
$ | 602 | | |
$ | 601 | |
| Catamaran tray sets | |
| 538 | | |
| 193 | |
| IT equipment | |
| 56 | | |
| 56 | |
| Leasehold improvements | |
| 15 | | |
| - | |
| Lab equipment | |
| 14 | | |
| 14 | |
| Office furniture | |
| 9 | | |
| 9 | |
| Fixed assets, gross | |
| 1,234 | | |
| 873 | |
| Less: accumulated depreciation | |
| (273 | ) | |
| (80 | ) |
| Fixed assets, net | |
$ | 961 | | |
$ | 793 | |
Construction in progress is made up of reusable
components that will become Catamaran Tray Sets. Depreciation expense was approximately $193 and $78 for the years ended December 31,
2023 and 2022, respectively.
6. Accrued Expenses
Accrued expenses consisted of the following:
| | |
December 31,
2023 | | |
December 31,
2022 | |
| Accrued compensation | |
$ | 334 | | |
$ | 452 | |
| Other accrued expenses | |
| 474 | | |
| 265 | |
| Total accrued expenses | |
$ | 808 | | |
$ | 717 | |
7. Debt
Convertible notes payable
In November 2023, the Company entered into Securities
Purchase Agreements with certain investors (the “Investors”), pursuant to which the Company sold to the Investors a total
of $1,250,000 in secured notes (the “Convertible Notes”) and warrants to purchase 45,000 shares of the Company’s common
stock at an exercise price equal to $1.94 per share.
The Convertible Notes bear an interest rate of
10% per annum with a default rate of 12% per annum and have a maturity date of November 21, 2024. All principal and accrued interest is
payable at maturity. At any time during the term of the Convertible Notes, the principal amount together with all accrued interest thereon
(the “Prepayment Amount”) may be paid in full, but not in part, by the Company. The Prepayment Amount may be paid by the Company
in cash or by the issuance to the Investors of shares of Series A Preferred Stock, if prior to such payment with Series A Preferred Stock
(i) certain stockholder proposals described in the Convertible Notes are approved by the Company’s stockholders; and (ii) the Company
has commitments from investors other than the Investors to purchase shares of Series A Preferred Stock with a stated value of at least
$3,750,000. The Convertible Notes are secured by a first priority security interest in all of the assets of the Company. The warrants
expire five years from the issuance date. The warrants contain a “cashless exercise” feature and contain anti-dilution rights
on subsequent issuances of equity or equity equivalents.
On February 20, 2024, the Investors agreed to
a complete prepayment of the Company’s obligations under the Convertible Notes, including accrued interest, in exchange for 84,729
shares of Series A Preferred Stock and warrants to purchase 157,094 shares of our common stock at $1.2705 per share and the Convertible
Notes were cancelled. See Note 13.
8. Leases
In June 2021, the Company entered into a facility
lease agreement for its company headquarters in Los Gatos, California. This non-cancellable operating lease expires in June 2026.
Operating lease costs for the facility lease were
$292 and $292 for the years ended December 31, 2023 and 2022, respectively.
Supplemental balance sheet information related
to leases was as follows:
| | |
December 31,
2023 | | |
December 31,
2022 | |
| Operating lease right-of-use asset | |
$ | 646 | | |
$ | 873 | |
| | |
| | | |
| | |
| Operating lease liability, current | |
$ | (256 | ) | |
$ | (228 | ) |
| Operating lease liability, noncurrent | |
| (428 | ) | |
| (683 | ) |
| Total operating lease liabilities | |
$ | (684 | ) | |
$ | (911 | ) |
Future maturities of operating lease liabilities
as of December 31, 2023 were as follows:
| 2024 | |
| 302 | |
| 2025 | |
| 310 | |
| 2026 | |
| 144 | |
| Total lease payments | |
| 756 | |
| Less: imputed interest | |
| (72 | ) |
| Present value of operating lease liabilities | |
$ | 684 | |
Other information:
| Cash paid for operating leases for the year ended December 31, 2023 | | $ | 293 | |
| Cash paid for operating leases for the year ended December 31, 2022 | | $ | 284 | |
| Remaining lease term - operating leases (in years) | | | 2.50 | |
| Average discount rate - operating leases | | | 8.0 | % |
9. Stockholders’ Equity
The Company’s current Amended and Restated
Certificate of Incorporation dated February 18, 2014 authorizes the issuance of 130,000,000 shares of common stock and 20,000,000 shares
of preferred stock, both with a par value of $0.001 per share. With respect to the preferred stock, 4,500,000 shares are designated Series
A Preferred Stock and 491,222 shares are designated Series B Preferred Stock. As of December 31, 2023 and 2022, there were no shares of
Series A Preferred stock or Series B Preferred Stock issued and outstanding.
Initial Public Offering
On April 26, 2022, the Company’s Registration
Statement relating to the IPO was declared effective by the SEC. The IPO consisted of 320,000 shares of common stock, par value $0.001
per share at a public offering price of $50.00 per share. Pursuant to the Underwriting Agreement dated April 26, 2022, between the Company,
The Benchmark Company, LLC (“Benchmark”) and Valuable Capital Limited (together with Benchmark, the “Underwriters”),
the Company granted the Underwriters warrants to purchase a total of 9,600 shares of the Company’s common stock at an exercise price
of $50.00 per share. The warrants expire on the fifth anniversary of the commencement of sales under the IPO. On April 27, 2022, the shares
of the Company’s common stock began trading on the Nasdaq Capital Market LLC under the symbol “TNON.”
On April 29, 2022, the IPO closed, and the Company
received approximately $13.8 million in net proceeds from the IPO after deducting the underwriting discount and commission and other estimated
IPO expenses payable by the Company. As a result of the completion of the IPO, the Company converted the entirety of the outstanding principal
and accrued interest of the convertible notes payable to 395,542 shares of the Company’s common stock.
On April 29, 2022, as result of the completion
of the IPO, the Company converted all shares of Series A and Series B Preferred Stock to 269,334 shares of the Company’s common
stock at the conversion rate detailed below and issued the common stock to the preferred stockholders.
Concurrent with the completion of the IPO and
in accordance with the Amended and Restated Exclusive Sales Representative Agreement executed in May 2021, the counterparty to the agreement
received anti-dilution protections to maintain ownership of 3.0% of the fully diluted equity of the Company through the date of an initial
public offering and was issued 31,235 shares of the Company’s common stock to the Representative, fully satisfying the Company’s
obligations. Also, as a result of the completion of the IPO, the Company issued 8,574 shares of its common stock to a consultant. The
value of these shares issued at the IPO price of $50.00 per share was charged to operating expenses in the Company’s consolidated
financial statements.
Registered Offering
On June 16, 2023, the Company closed the Registered
Offering of a total of 1,000,000 units (the “Units”) for proceeds, net of issuance costs, of $4,808, with each Unit consisting
of (i) one share of the Company’s common stock, and (ii) two warrants, each warrant to purchase one share of the Company’s
common stock at an exercise price equal to $5.60 per share (the “Offering Warrants”). The Offering Warrants were exercisable
upon issuance and will expire five years from the date of issuance. Per the terms of the Offering Warrants, the exercise price reset on
July 16, 2023 to $3.146 per share.
At-the-Market Offering Program
On May 4, 2023, the Company entered into an Equity
Distribution Agreement to establish an at-the-market offering program, under which the Company may sell from time to time, at its option,
shares of its common stock having an aggregate gross sales price of $5.5 million. The Company is required to pay the Sales Agents a commission
of 3% of the gross proceeds from the sale of shares and has also agreed to provide the Sales Agents with customary indemnification rights.
During the year ended December 31, 2023, 232,100 shares of the Company’s common stock were sold under the program at a weighted-average
price of $2.27 per share with aggregate net proceeds of $495.
Equity Line of Credit
On July 24, 2023, the Company entered into a purchase
agreement (“Purchase Agreement”) with Lincoln Park Capital Fund, LLC (“Lincoln Park”), under which, subject to
specified terms and conditions, the Company may sell to Lincoln Park up to $10 million of shares of common stock from time to time during
the term of the Purchase Agreement. On September 22, 2023 (the “Commencement Date”), the Company filed a registration statement
with the Securities and Exchange Commission (the “SEC”), covering the resale of shares of common stock issued to Lincoln Park
under the Purchase Agreement.
Beginning on the Commencement Date and for a period
of 24 months thereafter, under the terms and subject to the conditions of the Purchase Agreement, from time to time, at the Company’s
discretion, the Company has the right, but not the obligation, to sell to Lincoln Park, and Lincoln Park is obligated to purchase, up
to $10 million of shares of common stock, subject to certain limitations set forth in the Purchase Agreement. Specifically, from time
to time from and after the Commencement Date, the Company may, at its discretion, direct Lincoln Park to purchase on any single business
day on which the closing price of its common stock on The Nasdaq Capital Market (“Nasdaq”) is equal to or greater than $1.50
up to 10,000 shares of common stock (a “Regular Purchase”); provided, that the Company may direct Lincoln Park to purchase
in a Regular Purchase (i) up to 12,500 shares of common stock, if the closing sale price of its common stock on Nasdaq on such business
day is at least $15.00 per share and (ii) up to 15,000 shares of common stock, if the closing sale price of its common stock on Nasdaq
on such business day is at least $25.00 per share. In no case, however, will Lincoln Park’s commitment with respect to any single
Regular Purchase exceed $500,000; provided, that the parties may mutually agree at any time to increase the maximum number of shares of
common stock the Company may direct Lincoln Park to purchase in any single Regular Purchase to up to 100,000 shares or any number of shares
that shall not exceed 4.99% of the then outstanding shares of common stock. The foregoing share amounts and per share prices will be adjusted
for any reorganization, recapitalization, non-cash dividend, stock split, reverse stock split or other similar transaction occurring after
the date of the Purchase Agreement with respect to our common stock. The purchase price per share for each such Regular Purchase will
be based on prevailing market prices of the Company’s common stock immediately preceding the time of sale, as determined under the
Purchase Agreement.
Voting rights
The holders of vested shares of common stock are
entitled to vote on any matter submitted to a vote of the stockholders and each such holder is entitled to one vote per share of common
stock held. The holders of Series A and Series B Preferred Stock were entitled to vote together with the common stock as a single class
on any matter submitted to a vote of the stockholders. Holders of Series A and Series B Preferred Stock were entitled to the number of
votes equal to the number of common stock issuable upon conversion of their respective Series A and Series B Preferred Stock at the time
such shares are voted. The holders of a majority of the preferred stock had additional voting rights as specified in the Company’s
Amended and Restated Certificate of Incorporation, as amended.
Equity awards
In 2012, the Board of Directors of the Company
(the “Board”) approved the Tenon Medical, Inc. 2012 Equity Incentive Plan (the “2012 Plan”). The 2012 Plan provided
for the issuance of common stock options, appreciation rights, and other awards to employees, directors, and consultants. Options issued
under the 2012 Plan generally vest over a period of two to four years and have a 10-year expiration date.
On January 10, 2022 and February 2, 2022, the
Board and stockholders, respectively, of the Company approved the Tenon Medical, Inc. 2022 Equity Incentive Plan (the “2022 Plan”),
which was effective on April 25, 2022. The initial number of shares of common stock subject to awards under the 2022 Plan was 160,000.
The 2022 Plan calls for automatic annual increases in the number of shares available for issuance equal to the least of (a) 110,000 shares,
(b) 4% of the total number of shares of all classes of common stock outstanding on the last day of the immediately preceding fiscal year,
or (c) such number determined by the 2022 Plan administrator no later than the last day of the immediately preceding fiscal year. Annual
increases will continue until the tenth anniversary of the earlier of the Board or stockholder approval of the 2022 Plan, which is January
10, 2032. Upon the effective date of the 2022 Plan, the Board terminated the 2012 Plan such that no new equity awards will be issued by
the 2012 Plan.
Compensation expense for the years ended December
31, 2023 and 2022 includes the portion of awards vested in the periods for all equity-based awards granted, based on the grant date fair
value. estimated using a Black-Scholes option valuation model. Grant date fair value for restricted stock units is estimated using the
fair value of the Company’s common stock on the date of grant. Grant date fair value for stock options is estimated using a Black-Scholes
option valuation model using the weighted-average assumptions in the table below:
| | | Years ended
December 31, | |
| | | 2023 | | | 2022 | |
| Expected volatility | | | 63.89 | % | | | 57.68 | % |
| Dividend yield | | | 0 | % | | | 0 | % |
| Risk-free interest rate | | | 4.28 | % | | | 3.34 | % |
| Expected term in years | | | 5.85 | | | | 5.85 | |
Estimates of fair value are not intended to predict
actual future events or the value ultimately realized by employees who receive equity awards, and subsequent events are not indicative
of the reasonableness of the original estimates of fair value made by the Company in accordance with authoritative guidance.
A summary of the Company’s share option
and restricted stock unit activity under its plans is as follows:
| | | Options | | | RSUs | |
| | | Number of
Options | | | Weighted-
Average
Exercise
Price per Share | | | Weighted-
Average
Remaining
Contractual
Term
(In Years) | | | Number of
RSUs | | | Weighted
Average Grant
Date Fair
Value per
Share | |
| Balance as of December 31, 2021 | | | 72,744 | | | $ | 53.18 | | | | 7.12 | | | | - | | | | | |
| Granted | | | 17,145 | | | $ | 22.98 | | | | | | | | 131,858 | | | $ | 79.29 | |
| Balance as of December 31, 2022 | | | 89,889 | | | $ | 47.42 | | | | 8.10 | | | | 131,858 | | | $ | 79.29 | |
| Granted | | | 15,050 | | | $ | 12.91 | | | | | | | | 7,500 | | | $ | 2.91 | |
| Released | | | - | | | | - | | | | | | | | (61,200 | ) | | $ | 82.04 | |
| Canceled | | | (2,850 | ) | | $ | 39.87 | | | | | | | | (1,242 | ) | | $ | 88.60 | |
| Balance as of December 31, 2023 | | | 102,089 | | | $ | 42.54 | | | | 7.41 | | | | 76,916 | | | $ | 69.50 | |
| Exercisable at December 31, 2023 | | | 70,634 | | | $ | 48.66 | | | | 6.86 | | | | | | | | | |
The weighted-average grant-date fair value of
options granted during the years ended December 31, 2023 and 2022 was $7.63 and $12.90, respectively. The aggregate intrinsic value of
outstanding options at December 31, 2023 was $0. The aggregate intrinsic value is equal to the difference between the exercise price of
the underlying option and the fair value of the Company’s common stock for in-the-money options. As of December 31, 2023, total
compensation cost not yet recognized related to unvested options was $414, which is expected to be recognized over a weighted-average
period of 0.99 years, and total compensation costs not yet recognized related to unvested RSUs was $4,773, which is expected to be recognized
over a weighted-average period of 1.40 years.
The following table sets forth stock-based compensation
expense recognized for the years ended December 31, 2023 and 2022:
| | |
Years ended
December 31, | |
| | |
2023 | | |
2022 | |
| Research and development | |
$ | 1,504 | | |
$ | 995 | |
| Sales and marketing | |
| 217 | | |
| 117 | |
| General, and administrative | |
| 2,424 | | |
| 1,785 | |
| Total stock-based compensation expense | |
$ | 4,145 | | |
$ | 2,897 | |
At December 31, 2023, there were 37,486 shares
available for issuance under the 2022 Plan.
Warrants
In April 2022, as noted above, the Company granted
the Underwriters warrants to purchase a total of 9,600 shares of the Company’s common stock. The warrants are immediately exercisable
at an exercise price of $50.00 per share and expire on the fifth anniversary of the commencement of sales under the IPO. The fair value
of the warrants on the grant date was $27.50 per warrant, which was calculated using a Black-Scholes option valuation model with an expected
term of 5.00 years, expected volatility of 62.55%, dividend yield of 0%, and risk-free interest rate of 2.92%. The Company recorded the
fair value of these warrants of approximately $264 as an issuance cost to additional paid-in capital in 2022. As the IPO issuance costs
were also recorded to additional paid-in capital, the net impact was $0.
In June 2023, as noted above, in connection with
the Registered Offering, the Company issued Offering Warrants to purchase a total of 2,000,000 shares of the Company’s common stock.
The Offering Warrants were exercisable upon issuance at an exercise price of $5.60 per share and will expire five years from the date
of issuance. Per the terms of the Offering Warrants, the exercise price of the Offering Warrants reset on July 16, 2023, to a price equal
to the greater of (i) $2.80 per share and (ii) 100% of the last VWAP (as defined in the Warrants) on July 14, 2023, which was $3.146 per
share. The fair value of the Offering Warrants on the grant date was approximately $3,164, or $1.58 per warrant, which was calculated
using a Monte-Carlo simulation to estimate the final exercise price, which is considered a Level 3 fair value measurement, using as inputs;
the starting value of $3.00 per share, the Company’s VWAP on June 16; an assumed daily distribution of returns; a mean daily return
of 5.18%; a short-term annual volatility of 100% and a standard deviation of 6.3%. The model used Black-Scholes to then calculate the
estimated fair value of the Offering Warrants, using an estimated time to maturity of 4.9 years, a risk-free interest rate of 3.99% and
a long-term volatility of 60%. Based on the accounting guidance under ASC 815, the Company determined that the Offering Warrants did not
meet the criteria for classification as equity as of June 30, 2023. Accordingly, the Company classified the fair value of the Offering
Warrants as a liability. As of July 16, 2023, with the resolution of the reset value, the Company has determined that the Offering Warrants
do meet the criteria for classification as equity and the fair value of the Offering Warrants has been reclassified to additional paid-in
capital on the Company’s consolidated balance sheet as of that date.
In November 2023, in connection with the issuance
of the Convertible Notes, the Company issued warrants to purchase a total of 45,000 shares of the Company’s common stock at an exercise
price equal to $1.94 per share. The warrants expire five years from the issuance date. The fair value of the warrants on the grant date
was $1.29 per warrant, which was calculated using a Black-Scholes option valuation model with an expected term of 5.00 years, expected
volatility of 68.89%, dividend yield of 0%, and risk-free interest rate of 4.41%. The Company recorded the fair value of these warrants
of approximately $58 as an issuance cost to additional paid-in capital in 2023.
10. Commitments and Contingencies
Sales Representative Agreement
In April 2020, the Company entered into an Exclusive
Sales Representative Agreement, under which the counterparty to the agreement (the “Representative”) received exclusive rights
to market, promote, and distribute The Catamaran System in the United States and Puerto Rico. The agreement is for an initial period of
five years, and automatically renews for an additional five years unless written notice is given by either party prior to April 27, 2023.
The agreement provides for a bonus to be paid to the Representative upon an acquisition or IPO. In May 2021, the Company entered into
an Amended and Restated Exclusive Sales Representative Agreement (the “Restated Sales Agreement”). In connection with the
amended agreement, the Company paid $500 cash and issued 53,757 shares of common stock to the Representative, for which the Company recorded
a combined total of approximately $880 as sales and marketing expense. In addition, the Representative received anti-dilution protections
to maintain ownership of 3.0% of the fully diluted equity of the Company through the date of an initial public offering. In October 2021,
the Company issued 4,445 shares of common stock with a fair value of approximately $333 to the Representative in accordance with the anti-dilution
provision. In April 2022, the Company issued 31,235 shares of common stock to the Representative in accordance with the anti-dilution
provision, fully satisfying the Company’s obligations.
The Restated Sales Agreement restructured the
calculation of the bonus paid to the Representative upon an acquisition, removed the bonus payable upon an IPO, and allows the Company
to terminate the Restated Sales Agreement as long as the bonus paid to the Representative is at least $6,000.
On October 6, 2022, the Company entered into the
Terminating Amended and Restated Exclusive Sales Representative Agreement (the “Termination Agreement”) with the Representative,
which terminated the Restated Sales Agreement. In accordance with the Termination Agreement, (i) the Company paid the Representative $1,000
in cash; and (ii) the Company agreed to pay the Representative (a) $85 per month during the six months after the date of the Termination
Agreement in return for efforts by the Representative to transition operations to the Company, (b) 20% of net sales of the product sold
in the United States and Puerto Rico until December 31, 2023 and (c) after December 31, 2023, 10% of net sales until such time as the
aggregate amount paid to the Representative under this clause (c) and clause (b) above equal $3,600. In the event of an acquisition of
the Company, the Company will pay the Representative $3,600 less previous amounts paid pursuant to clause (b) and clause (c) above. The
Company recorded a charge of $1,000 for the payment to the Representative in the fourth quarter of 2022 and expensed the $85 per month
charges as incurred over the six-month period. For payments under clause (b) and clause (c) above, the Company estimated the fair value
of the liability using level 3 hierarchy inputs based on a Monte Carlo simulation of future revenues with a 25% quarterly estimated standard
deviation of growth rates and a 10% probability of dissolution, discounted at an estimated discount rate of 15.4%. Based on the Company’s
fair value analysis, a total of $2,611 was charged to sales and marketing expense in the consolidated statements of operations and comprehensive
loss and recorded as accrued commissions in the consolidated balance sheets. A reconciliation of the liability under clause (b) and clause
(c) for the year ended December 31, 2023 is as follows:
| | |
2023 | |
| Balance at January 1, 2023 | |
$ | 2,560 | |
| Amounts paid during 2023 | |
| (592 | ) |
| Accretion | |
| 409 | |
| Balance at December 31, 2023 | |
$ | 2,377 | |
Per the terms of the Termination Agreement, the
Company ultimately expects to expense $3,600 under clause (b) and clause (c).
Simultaneously with the execution of the Termination
Agreement, the Company entered into a Consulting Agreement dated October 6, 2022, with the Representative (the “Consulting Agreement”).
Under the terms and conditions of the Consulting Agreement, the Representative is tasked with organizing, recruiting, training, and coordinating
the Company’s Clinical Specialist program, Physician Education program and Sales Education program as more specifically described
in the Consulting Agreement.
The term of the Consulting Agreement was from
October 6, 2022, until October 5, 2023, when it terminated in accordance with the terms of the Consulting Agreement. In consideration
for the services to be provided, the Company paid the Representative a base consulting fee of $700 per year, payable in monthly instalments,
along with additional compensation of $62.5 per quarter, if certain sales targets were met, for four quarters; along with any travel and
related out-of-pocket expenses incurred by the Representative in connection with the performance of the services.
Litigation
In the normal course of business, the Company
may possibly be named as a defendant in various lawsuits.
11. Concentrations of Risk
Credit risk
Financial instruments that potentially subject
the Company to concentrations of credit risk consist principally of cash and cash equivalents.
The Company maintains cash balances at financial
institutions located in California and Switzerland. Accounts at the U.S. financial institutions are secured by the Federal Deposit Insurance
Corporation. At times, balances may exceed federally insured limits. The Company has not experienced any losses in such accounts. Management
believes that the Company is not exposed to any significant credit risk with respect to its cash and cash equivalents.
The Company grants unsecured credit to its customers
based on an evaluation of the customer’s financial condition and a cash deposit is generally not required. Management believes its
credit policies do not result in significant adverse risk and historically has not experienced significant credit-related losses.
Currency risk
The Company’s subsidiary, Tenon Technology
AG, realizes a portion of its expenses in Swiss francs. Consequently, certain assets and liabilities are exposed to foreign currency fluctuations.
At December 31, 2023 and 2022, approximately $741 and $8, respectively, of the Company’s net monetary assets were denominated in
Swiss francs. The Company has not entered into any hedging transactions to reduce the exposure to currency risk.
12. Income Taxes
The components of loss before income taxes are
as follows:
| | |
Years ended
December 31, | |
| | |
2023 | | |
2022 | |
| United States | |
$ | (15,570 | ) | |
$ | (18,886 | ) |
| International | |
| (11 | ) | |
| (30 | ) |
| Loss before income taxes | |
$ | (15,581 | ) | |
$ | (18,916 | ) |
The components of current income tax expense are
as follows:
| | |
Years ended
December 31, | |
| | |
2023 | | |
2022 | |
| Federal | |
$ | - | | |
$ | - | |
| State | |
| - | | |
| 1 | |
| Foreign | |
| - | | |
| - | |
| Total income tax expense | |
$ | - | | |
$ | 1 | |
A reconciliation of the expected tax computed
at the U.S. statutory federal income tax rate to the total provision for income taxes for the years ended December 31, 2023 and 2022 is
as follows:
| | |
Years ended
December 31, | |
| | |
2023 | | |
2022 | |
| Statutory rate | |
| (21 | )% | |
| (21 | )% |
| State taxes, net of federal benefit | |
| (7 | )% | |
| (7 | )% |
| Non-deductible differences | |
| 3 | % | |
| 1 | % |
| Change in valuation allowance | |
| 25 | % | |
| 27 | % |
| Provision for taxes | |
| - | | |
| - | |
Significant components of the Company’s
net deferred tax assets at December 31, 2023 and 2022 are as follows:
| | |
Years ended
December 31, | |
| | |
2023 | | |
2022 | |
| Deferred tax assets: | |
| | |
| |
| Net operating loss carryforwards | |
$ | 9,504 | | |
$ | 7,001 | |
| Credit carryforwards | |
| 220 | | |
| 109 | |
| Fixed assets | |
| 52 | | |
| - | |
| Accruals and reserves | |
| 111 | | |
| 126 | |
| Stock-based compensation | |
| 1,802 | | |
| 843 | |
| Intangibles | |
| 220 | | |
| 244 | |
| Operating lease liability | |
| 188 | | |
| 254 | |
| Capitalized research and development | |
| 514 | | |
| 274 | |
| Total deferred tax assets | |
| 12,611 | | |
| 8,851 | |
| Valuation allowance | |
| (12,433 | ) | |
| (8,564 | ) |
| Net deferred tax assets | |
| 178 | | |
| 287 | |
| Deferred tax liabilities: | |
| | | |
| | |
| Fixed assets | |
| - | | |
| (44 | ) |
| Operating lease right of use | |
| (178 | ) | |
| (243 | ) |
| Total deferred tax liabilities | |
| (178 | ) | |
| (287 | ) |
| Net deferred tax assets | |
$ | - | | |
| - | |
In assessing the realizability of deferred tax
assets at December 31, 2023, management considered whether it is more likely than not that some portion or all of the deferred tax assets
will be realized, and determined that a valuation allowance was required for those deferred tax assets that are not expected to provide
future tax benefits. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during
the periods in which those temporary differences become deductible.
At December 31, 2023, the Company has available
net operating loss carryforwards of approximately $33,866 for federal income tax purposes, of which approximately $33,644 was generated
after 2017 and can be carried forward indefinitely under the Tax Cuts and Jobs Act. The remaining federal net operating loss of approximately
$222, which was generated prior to 2018, will start to expire in 2034 if not utilized.
At December 31, 2023, the net operating loss carryforwards
for state purposes are approximately $32,147 and will begin to expire in 2032 if not utilized. In addition, the Company had foreign net
operating loss carryforwards of approximately $1,378 at December 31, 2023 that will start to expire in 2024 if not utilized.
The Company had credit carryforwards of approximately
$214 for federal income tax purposes. The federal tax credits will begin to expire in 2041.
The Company also had credit carryforwards of approximately
$101 for California income tax purposes. These credits have no expiration.
The Company has not completed a study to determine
whether any ownership change per the provisions of Section 382 of the Internal Revenue Code of 1986, as amended, as well as similar state
provisions, has occurred. Utilization of the Company’s net operating loss and income tax credit carryforwards may be subject to
a substantial annual limitation due to ownership changes that may have occurred or that could occur in the future. These ownership changes
may limit the amount of the net operating loss and income tax credit carryover that can be utilized annually to offset future taxable
income. In general, an “ownership change” as defined by Section 382 of the Code results from a transaction or series of transactions
over a three-year period resulting in an ownership change of more than 50 percentage points of the outstanding stock of a company by certain
stockholders.
Uncertain tax positions
In accordance with authoritative guidance, the
impact of an uncertain income tax position on the income tax return must be recognized at the largest amount that is more likely than
not to be sustained upon audit by the relevant taxing authority. An uncertain income tax position will not be recognized if it has less
than a 50% likelihood of being sustained. The following shows the changes in the gross amount of recognized tax benefits:
| | |
Years ended
December 31, | |
| | |
2023 | | |
2022 | |
| Unrecognized tax benefits, beginning of year | |
$ | 38 | | |
$ | - | |
| Increases related to prior year tax positions | |
| 5 | | |
| 12 | |
| Decreases related to prior year tax positions | |
| - | | |
| - | |
| Increases related to current year tax positions | |
| 36 | | |
| 26 | |
| Unrecognized tax benefits, end of year | |
$ | 79 | | |
$ | 38 | |
The Company recognizes interest and penalties
related to unrecognized tax positions within the income tax expense line in the accompanying consolidated statements of operations and
comprehensive loss. The Company does not anticipate that its total unrecognized tax benefits will significantly change due to settlement
of examination or the expiration of statute of limitations during the next 12 months. Due to the full valuation allowance at December
31, 2023, current adjustments to the unrecognized tax benefit will have no impact on our effective income tax rate.
The Company currently has no federal or state
tax examinations in progress nor has it had any federal or state tax examinations since its inception. As a result of the Company’s
net operating loss and credit carryforwards, all of its years are subject to federal and state examination.
13. Subsequent Events
On February 20, 2024, the Company entered into
a Securities Purchase Agreement (the “Purchase Agreement”) with certain investors, pursuant to which the Company agreed to
sell, issue and deliver to these investors, in a private placement offering (the “Offering”), a total of 172,239 shares of
the Company’s Series A Preferred Stock and warrants (the “Warrants”) to purchase 258,374 shares of common stock, par
value $0.001 per share, of the Company (“Common Stock”) at an exercise price equal to $1.2705 per share for an aggregate offering
price of $2,605,000.
Additionally, on February 20, 2024, the Investors
agreed to a complete prepayment of the Company’s obligations under the Convertible Notes, including accrued interest, in exchange
for 84,729 shares of Series A Preferred Stock and warrants to purchase 157,094 shares of our common stock at $1.2705 per share and the
Convertible Notes were cancelled. The Warrants are immediately exercisable and expire five years from the date of issuance.
The Series A Preferred Stock is convertible, at
any time, at the option of the holder into shares of Common Stock. Each share of Series A Preferred Stock shall be convertible, at any
time after the date of issuance, at the option of the holder thereof (or, upon a Required Conversion (as defined below), at the option
of the Corporation), into that number of shares of Common Stock determined by dividing the Stated Value (as defined below) for such share
of Series A Preferred Stock by the Conversion Price (as defined below). “Stated Value” means for any share of Series A Preferred
Stock, an amount equal to the product of (x) $15.125 multiplied by (y) the sum of 1 plus the product of (A) 0.06 multiplied by (B) a fraction
equal to the number of days that such share of Series A Preferred Stock has been issued divided by 365. “Conversion Price”
means (i) for the shares of Series A Preferred Stock issued on the Closing Date, $1.5125 and (ii) for each share of Series A Preferred
Stock issued thereafter, an amount equal to the greater of (x) $1.5125 and the average of the VWAPs for the 10 Trading Days prior the
issuance date of such share of Series A Preferred Stock, in each case subject to adjustment as set forth herein. On any date that ten
out of the last 15 daily VWAPs of the Common Stock is 250% higher than the Conversion Price on such date, then the Company will have the
right to require 50% of the Preferred Stock to be converted into shares of Common Stock. Additionally, on and after the time on which
the Company has $2.25 million in revenues in any single financial quarter, the Company will have the right to require 50% of the Preferred
Stock to be converted into shares of Common Stock (a “Required Conversion”). No dividends are payable on the Series A Preferred
Stock. The Series A Preferred Stock will vote together with the Common Stock on all matters other than as required by law; provided however
that any additional shares underlying the Series A Preferred Stock as a result of the anti-dilution provision described below shall not
vote on an “as converted” basis and shall only vote when issued upon conversion. Notwithstanding the foregoing, the vote of
an individual holder of Series A Preferred Stock (and underlying Common Stock) shall be capped at 9.99% (or 4.99% if selected by the holder).
The Conversion Price is subject to anti-dilution
adjustment as the result of any subdivision, combination of shares or recapitalization, stock dividends, stock splits and similar transactions
affecting the Common Stock. In addition, the Series A Preferred Stock will have weighted average anti-dilution protection providing for
adjustment of the Conversion Price in the event of issuance of, or commitments to issue, Common Stock for less than the Conversion Price
then in effect immediately prior to such issue or sale (a “Dilutive Issuance”), subject to customary exceptions; provided
however the anti-dilution for Dilutive Issuances shall not be operative until the stockholders of the Company have approved the terms
of the Series A Preferred Stock. Upon any liquidation or winding up of the Company (a “Liquidation”), the holders of Series
A Preferred Stock will be entitled to receive in preference to any other class or series of the Company’s equity securities the
greater of (i) the Stated Value plus accrued and unpaid dividends and (ii) what would be paid if the Series A Preferred Stock plus accrued
and unpaid dividends had been converted into Common Stock. A consolidation or merger of the Company or sale or transfer of all or substantially
all of its assets, or any transaction which results in the stockholders of the Company owning less than 50% of the equity or voting power
of the surviving entity (excluding the issuance of Common Stock in any financing transaction unless more than 50% of the Company’s
shares are issued to one stockholder or a number of stockholders who act as a one group) shall be deemed a Liquidation (a “Deemed
Liquidation”) with respect to the shares of Series A Preferred Stock of any holder who opts to have such occurrence treated as a
Deemed Liquidation; provided that if the liquidation preference payable on a Deemed Liquidation is less than 110% of the stated value
of the Series A Preferred Stock, the dividend rate on any accrued and unpaid dividends payable with respect to such Deemed Liquidation
will increase to 10%. All liquidation preferences payable in respect of a Deemed Liquidation will be payable in shares of Common Stock
based on the closing price of the Common Stock on the date of such Deemed Liquidation. Consent of the majority of the holders will be
required to (i) amend the Certificate of Incorporation or Bylaws of the Company so as to adversely alter the rights, preferences, privileges
of the Series A Preferred Stock, (ii) create any new class of shares pari passu or senior to the Series A Preferred Stock or increase
or decrease the number of authorized shares of Common Stock or preferred stock, (iii) pay or declare any dividend on Common Stock or other
junior securities, or incur indebtedness in any single transaction in excess of $1 million or (iv) redeem, purchase or otherwise acquire
any share or shares of preferred stock or Common Stock (other than (a) the repurchase of shares of Common Stock pursuant to a written
benefit plan or employment or consulting agreement, or (b) the repurchase of any equity securities in connection with the Company’s
right of first offer with respect to those securities contained in any written agreement with the Company).
As of March 29, 2024, with the issuance of the
Series A Preferred Stock, the conversion of the Convertible Notes, and proceeds from the Company’s ATM and ELOC facilities, the
Company believes that its Stockholders’ Equity will exceed $2.5 million and will therefore meet the minimum stockholder equity amount
required by the Nasdaq Stock Market, LLC.
Tenon Medical, Inc.
Condensed Consolidated
Balance Sheets (Unaudited)
(In thousands, except
share data)
| | |
June 30, | | |
December 31, | |
| | |
2024 | | |
2023 | |
| Assets | |
| | |
| |
| Current assets: | |
| | |
| |
| Cash and cash equivalents | |
$ | 1,968 | | |
$ | 2,428 | |
| Accounts receivable, net | |
| 679 | | |
| 518 | |
| Inventory | |
| 609 | | |
| 554 | |
| Prepaid expenses and other current assets | |
| 831 | | |
| 389 | |
| Total current assets | |
| 4,087 | | |
| 3,889 | |
| Fixed assets, net | |
| 904 | | |
| 961 | |
| Deposits | |
| 51 | | |
| 51 | |
| Operating lease right-of-use asset | |
| 525 | | |
| 646 | |
| Deferred offering costs | |
| 439 | | |
| 798 | |
| TOTAL ASSETS | |
$ | 6,006 | | |
$ | 6,345 | |
| | |
| | | |
| | |
| Liabilities and Stockholders’ EQUITY | |
| | | |
| | |
| Current liabilities: | |
| | | |
| | |
| Accounts payable | |
$ | 1,111 | | |
$ | 433 | |
| Accrued expenses | |
| 932 | | |
| 808 | |
| Current portion of accrued commissions | |
| 1,089 | | |
| 470 | |
| Current portion of operating lease liability | |
| 271 | | |
| 256 | |
| Convertible notes payable and accrued interest, net of debt discount of $0 and $77 at June 30, 2024 and December 31, 2023, respectively | |
| — | | |
| 1,173 | |
| Total current liabilities | |
| 3,403 | | |
| 3,140 | |
| Accrued commissions, net of current portion | |
| 1,481 | | |
| 1,999 | |
| Operating lease liability, net of current portion | |
| 290 | | |
| 428 | |
| Total liabilities | |
| 5,174 | | |
| 5,567 | |
| Commitments and contingencies (Note 8) | |
| | | |
| | |
| Stockholders’ equity: | |
| | | |
| | |
| Series A convertible preferred stock, $0.001 par value; 4,500,000 shares authorized at June 30, 2024 and December 31, 2023; 256,968 and 0 shares issued and outstanding at June 30, 2024 and December 31, 2023, respectively | |
| 3,300 | | |
| — | |
| Common stock, $0.001 par value; 130,000,000 shares authorized at June 30, 2024 and December 31, 2023; 3,780,827 and 2,600,311 shares issued and outstanding at June 30, 2024 and December 31, 2023, respectively | |
| 4 | | |
| 3 | |
| Additional paid-in capital | |
| 60,003 | | |
| 55,894 | |
| Accumulated deficit | |
| (62,475 | ) | |
| (55,073 | ) |
| Accumulated other comprehensive loss | |
| — | | |
| (46 | ) |
| Total stockholders’ equity | |
| 832 | | |
| 778 | |
| TOTAL LIABILITIES AND STOCKHOLDERS' EQUITY | |
$ | 6,006 | | |
$ | 6,345 | |
The accompanying notes are an integral part
of these condensed consolidated financial statements.
Tenon Medical, Inc.
Condensed Consolidated Statements of Operations
and Comprehensive Loss (Unaudited)
(In thousands, except per share data)
| | |
Three Months Ended
June 30, | | |
Six Months Ended
June 30, | |
| | |
2024 | | |
2023 | | |
2024 | | |
2023 | |
| Revenue | |
$ | 901 | | |
$ | 743 | | |
$ | 1,620 | | |
$ | 1,176 | |
| Cost of sales | |
| 431 | | |
| 549 | | |
| 680 | | |
| 1,029 | |
| Gross Profit | |
| 470 | | |
| 194 | | |
| 940 | | |
| 147 | |
| | |
| | | |
| | | |
| | | |
| | |
| Operating Expenses | |
| | | |
| | | |
| | | |
| | |
| Research and development | |
| 708 | | |
| 901 | | |
| 1,377 | | |
| 1,735 | |
| Sales and marketing | |
| 1,448 | | |
| 1,883 | | |
| 2,829 | | |
| 3,909 | |
| General and administrative | |
| 2,186 | | |
| 1,732 | | |
| 4,112 | | |
| 3,711 | |
| Total Operating Expenses | |
| 4,342 | | |
| 4,516 | | |
| 8,318 | | |
| 9,355 | |
| | |
| | | |
| | | |
| | | |
| | |
| Loss from Operations | |
| (3,872 | ) | |
| (4,322 | ) | |
| (7,378 | ) | |
| (9,208 | ) |
| | |
| | | |
| | | |
| | | |
| | |
| Other Income (Expense) | |
| | | |
| | | |
| | | |
| | |
| Gain on investments | |
| 39 | | |
| 37 | | |
| 66 | | |
| 93 | |
| Interest expense | |
| — | | |
| — | | |
| (34 | ) | |
| — | |
| Other income (expense), net | |
| 7 | | |
| — | | |
| (56 | ) | |
| — | |
| Total Other Income (Expense), net | |
| 46 | | |
| 37 | | |
| (24 | ) | |
| 93 | |
| Net Loss | |
$ | (3,826 | ) | |
$ | (4,285 | ) | |
$ | (7,402 | ) | |
$ | (9,115 | ) |
| Net Loss Per Share of Common Stock | |
| | | |
| | | |
| | | |
| | |
Basic and diluted | |
$ | (1.02 | ) | |
$ | (3.28 | ) | |
$ | (2.24 | ) | |
$ | (7.50 | ) |
| | |
| | | |
| | | |
| | | |
| | |
| Weighted-Average Shares of Common Stock Outstanding | |
| | | |
| | | |
| | | |
| | |
Basic and diluted | |
| 3,749 | | |
| 1,305 | | |
| 3,301 | | |
| 1,215 | |
| | |
| | | |
| | | |
| | | |
| | |
| Consolidated Statements of Comprehensive Loss: | |
| | | |
| | | |
| | | |
| | |
| Net loss | |
$ | (3,826 | ) | |
$ | (4,285 | ) | |
$ | (7,402 | ) | |
$ | (9,115 | ) |
| Unrealized gain on investments | |
| — | | |
| 3 | | |
| — | | |
| 16 | |
| Foreign currency translation adjustment | |
| — | | |
| 13 | | |
| 46 | | |
| 12 | |
| Total comprehensive loss | |
$ | (3,826 | ) | |
$ | (4,269 | ) | |
$ | (7,356 | ) | |
$ | (9,087 | ) |
The accompanying notes are an integral part
of these condensed consolidated financial statements.
Tenon Medical, Inc.
Condensed Consolidated Statements of Convertible
Preferred Stock and Stockholders’ Equity (Unaudited)
(In thousands, except share data)
Three months ended June 30, 2024 and 2023:
| | |
Series A Convertible
Preferred Stock | | |
Common Stock | | |
Additional Paid-In | | |
Accumulated | | |
Accumulated Other Comprehensive | | |
| |
| | |
Shares | | |
Amount | | |
Shares | | |
Amount | | |
Capital | | |
Deficit | | |
Loss | | |
Total | |
| Balance at March 31, 2024 | |
| 256,968 | | |
$ | 3,300 | | |
| 3,726,974 | | |
$ | 4 | | |
$ | 58,969 | | |
$ | (58,649 | ) | |
$ | — | | |
$ | 3,624 | |
| Stock-based compensation expense | |
| — | | |
| — | | |
| — | | |
| — | | |
| 1,034 | | |
| — | | |
| — | | |
| 1,034 | |
| Release of restricted stock units | |
| — | | |
| — | | |
| 53,853 | | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | |
| Net Loss | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | | |
| (3,826 | ) | |
| — | | |
| (3,826 | ) |
| Balance at June 30, 2024 | |
| 256,968 | | |
$ | 3,300 | | |
| 3,780,827 | | |
$ | 4 | | |
$ | 60,003 | | |
$ | (62,475 | ) | |
$ | — | | |
$ | 832 | |
| | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | |
| Balance at March 31, 2023 | |
| — | | |
$ | — | | |
| 1,125,128 | | |
$ | 1 | | |
$ | 46,883 | | |
$ | (44,322 | ) | |
$ | (88 | ) | |
$ | 2,474 | |
| Stock-based compensation expense | |
| — | | |
| — | | |
| — | | |
| — | | |
| 1,054 | | |
| — | | |
| — | | |
| 1,054 | |
| Release of restricted stock units | |
| — | | |
| — | | |
| 37,247 | | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | |
| Issuance of common stock and warrants, net of issuance costs | |
| — | | |
| — | | |
| 1,000,000 | | |
| 1 | | |
| 1,643 | | |
| — | | |
| — | | |
| 1,644 | |
| Other comprehensive income | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | | |
| 16 | | |
| 16 | |
| Net loss | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | | |
| (4,285 | ) | |
| — | | |
| (4,285 | ) |
| Balance at June 30, 2023 | |
| — | | |
$ | — | | |
| 2,162,375 | | |
$ | 2 | | |
$ | 49,580 | | |
$ | (48,607 | ) | |
$ | (72 | ) | |
$ | 903 | |
Six months ended June 30, 2024 and 2023:
| | |
Series
A Convertible
Preferred Stock | | |
Common
Stock | | |
Additional
Paid-In
| | |
Accumulated | | |
Accumulated
Other
Comprehensive
| | |
| |
| | |
Shares | | |
Amount | | |
Shares | | |
Amount | | |
Capital | | |
Deficit | | |
Loss | | |
Total | |
| Balance at December
31, 2023 | |
| — | | |
$ | — | | |
| 2,600,311 | | |
$ | 3 | | |
$ | 55,894 | | |
$ | (55,073 | ) | |
$ | (46 | ) | |
$ | 778 | |
| Stock-based compensation
expense | |
| — | | |
| — | | |
| — | | |
| — | | |
| 2,052 | | |
| — | | |
| — | | |
| 2,052 | |
| Issuance of common stock,
net of issuance costs | |
| — | | |
| — | | |
| 1,123,439 | | |
| 1 | | |
| 1,803 | | |
| — | | |
| — | | |
| 1,804 | |
| Issuance of series A convertible
preferred stock, net of issuance costs | |
| 256,968 | | |
| 3,300 | | |
| — | | |
| — | | |
| 254 | | |
| — | | |
| — | | |
| 3,554 | |
| Release of restricted stock
units | |
| — | | |
| — | | |
| 57,077 | | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | |
| Other comprehensive income | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | | |
| 46 | | |
| 46 | |
| Net
loss | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | | |
| (7,402 | ) | |
| — | | |
| (7,402 | ) |
| Balance
at June 30, 2024 | |
| 256,968 | | |
$ | 3,300 | | |
| 3,780,827 | | |
$ | 4 | | |
$ | 60,003 | | |
$ | (62,475 | ) | |
$ | — | | |
$ | 832 | |
| | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | |
| Balance
at December 31, 2022 | |
| — | | |
$ | — | | |
| 1,123,680 | | |
$ | 1 | | |
$ | 45,843 | | |
$ | (39,492 | ) | |
$ | (100 | ) | |
$ | 6,252 | |
| Stock-based compensation
expense | |
| — | | |
| — | | |
| — | | |
| — | | |
| 2,094 | | |
| — | | |
| — | | |
| 2,094 | |
| Release of restricted stock
units | |
| — | | |
| — | | |
| 38,695 | | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | |
| Issuance of common stock
and warrants, net of issuance costs | |
| — | | |
| — | | |
| 1,000,000 | | |
| 1 | | |
| 1,643 | | |
| — | | |
| — | | |
| 1,644 | |
| Other comprehensive income | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | | |
| 28 | | |
| 12 | |
| Net
loss | |
| — | | |
| — | | |
| — | | |
| — | | |
| — | | |
| (9,115 | ) | |
| — | | |
| (9,115 | ) |
| Balance
at June 30, 2023 | |
| — | | |
$ | — | | |
| 2,162,375 | | |
$ | 2 | | |
$ | 49,580 | | |
$ | (48,607 | ) | |
$ | (72 | ) | |
$ | 903 | |
The accompanying notes are an integral part
of these condensed consolidated financial statements.
Tenon Medical, Inc.
Condensed Consolidated Statements of Cash
Flows (Unaudited)
(In thousands)
| | |
Six Months Ended
June 30, | |
| | |
2024 | | |
2023 | |
| Cash Flows from Operating Activities | |
| | |
| |
| Net loss | |
$ | (7,402 | ) | |
$ | (9,115 | ) |
| Adjustments to reconcile net loss to net cash used in operating activities: | |
| | | |
| | |
| Stock-based compensation expense | |
| 2,052 | | |
| 2,094 | |
| Depreciation and amortization | |
| 189 | | |
| 60 | |
| Provision for losses on accounts receivable | |
| 46 | | |
| — | |
| Amortization of operating right-of-use asset | |
| 121 | | |
| 111 | |
| Increase (decrease) in cash resulting from changes in: | |
| | | |
| | |
| Accounts receivable | |
| (207 | ) | |
| (373 | ) |
| Inventory | |
| (55 | ) | |
| (155 | ) |
| Prepaid expenses and other assets | |
| (442 | ) | |
| (325 | ) |
| Accounts payable | |
| 678 | | |
| 362 | |
| Accrued expenses | |
| 385 | | |
| 698 | |
| Operating lease liability | |
| (123 | ) | |
| (109 | ) |
| Net cash used in operating activities | |
| (4,758 | ) | |
| (6,752 | ) |
| | |
| | | |
| | |
| Cash Flows from Investing Activities | |
| | | |
| | |
| Sales of short-term investments | |
| — | | |
| 6,503 | |
| Purchases of short-term investments | |
| — | | |
| (493 | ) |
| Purchases of property and equipment | |
| (119 | ) | |
| (212 | ) |
| Net cash (used in) provided by investing activities | |
| (119 | ) | |
| 5,798 | |
| | |
| | | |
| | |
| Cash Flows from Financing Activities | |
| | | |
| | |
| Proceeds from issuance of Series A Convertible Preferred Stock | |
| 2,437 | | |
| — | |
| Proceeds from issuance of common stock | |
| 1,934 | | |
| — | |
| Proceeds from issuance of common stock and warrants, net of issuance costs | |
| | | |
| 4,808 | |
| Deferred offering costs | |
| — | | |
| (143 | ) |
| Net cash provided by financing activities | |
| 4,371 | | |
| 4,665 | |
| | |
| | | |
| | |
| Effect of foreign currency translation on cash flow | |
| 46 | | |
| 12 | |
| Net (Decrease) Increase in Cash and Cash Equivalents | |
| (460 | ) | |
| 3,723 | |
| | |
| | | |
| | |
| Cash and Cash Equivalents at Beginning of Period | |
| 2,428 | | |
| 2,129 | |
| Cash and Cash Equivalents at End of Period | |
$ | 1,968 | | |
$ | 5,852 | |
| | |
| | | |
| | |
| Supplemental Disclosures of Cash Flow Information | |
| | | |
| | |
| Non-cash investing and financing activities: | |
| | | |
| | |
| Preferred stock issued upon conversion of debt and accrued interest | |
$ | 1,186 | | |
| — | |
| Reclassification of deferred offering costs to additional paid-in capital | |
$ | 130 | | |
| — | |
The accompanying notes are an integral part
of these condensed consolidated financial statements.
Notes to Condensed
Consolidated Financial Statements (unaudited) (in thousands, except share and per-share data)
1. Organization and Business
Nature of operations
Tenon Medical, Inc. (the “Company”)
was incorporated in the State of Delaware on June 19, 2012 and was headquartered in San Ramon, California until June 2021 when it relocated
to Los Gatos, California. The Company is a medical device company that has developed The Catamaran™ SI Joint Fusion System (“the
Catamaran System”) that offers a novel, less invasive approach to the sacroiliac joint (the “SI Joint”) using a single,
robust, titanium implant for treatment of the most common types of SI Joint disorders that cause lower back pain. The Company received
U.S. Food and Drug Administration (“FDA”) clearance in 2018 for The Catamaran System and is currently focused on the U.S.
market. Since the national launch of the Catamaran System in October 2022, the Company is focused on three commercial opportunities:
1) Primary SI Joint procedures, 2) Revision procedures of failed SI Joint implants and 3) SI Joint fusion adjunct to a spine fusion construct.
Principles of consolidation
The condensed consolidated financial statements
of the Company for the three and six months ended June 30, 2023 and as of December 31, 2023 include the accounts of its wholly-owned subsidiary,
Tenon Technology AG (“TTAG”), a Swiss company. All intercompany balances and transactions have been eliminated in consolidation.
The financial statements of TTAG are prepared for the same reporting period as the parent, using consistent accounting policies in all
material respects. In 2024, TTAG was effectively dissolved and, as such, the financial statements for the three and six months ended 2024
and as of June 30, 2024 only include the accounts of the Company.
2. Summary of Significant Accounting Principles
Basis of presentation
The accompanying unaudited condensed consolidated
financial statements have been prepared pursuant to the rules and regulations of the United States Securities and Exchange Commission
(the “SEC”). As permitted under these rules and regulations, the Company has condensed or omitted certain financial information
and footnote disclosures normally included in its annual consolidated financial statements prepared in accordance with accounting principles
generally accepted in the United States of America (“U.S. GAAP”). The condensed consolidated balance sheet as
of December 31, 2023 has been derived from the Company’s audited consolidated financial statements, which are included
in its Annual Report on Form 10-K filed with the SEC on March 29, 2024.
These unaudited condensed consolidated financial
statements and accompanying notes should be read in conjunction with the Company’s audited consolidated financial statements as
of and for the years ended December 31, 2023 and 2022 included in its Annual Report of Form 10-K filed with the SEC on March 29, 2024.
These condensed consolidated financial statements
have been prepared on the same basis as the Company’s annual consolidated financial statements and, in management’s opinion,
reflect all adjustments, consisting only of normal recurring adjustments, that are necessary for a fair presentation of its financial
information. The interim period operating results do not necessarily indicate the results that may be expected for any other interim
period or for the full fiscal year.
The Company’s significant accounting policies
are disclosed in the audited consolidated financial statements as of and for the years ended December 31, 2023 and 2022. There have been
no material changes in the Company’s significant accounting policies during the six months ended June 30, 2024.
Going concern uncertainty and liquidity requirements
The accompanying consolidated financial statements
have been prepared assuming the Company will continue as a going concern, which contemplates the realization of assets and the settlement
of liabilities and commitments in the normal course of business. There is substantial doubt about the Company’s ability to continue
as a going concern for one year after the date that these condensed consolidated financial statements are issued.
Since inception, the Company has incurred losses
and negative cash flows from operations. Management expects to incur additional operating losses and negative cash flows from operations
in the foreseeable future as the Company continues its product development programs and the commercialization of The Catamaran System.
Based on the Company’s expected level of revenues and expenditures, the Company believes that its existing cash and cash equivalents
as of June 30, 2024 will not provide sufficient funds to enable it to meet its obligations for a period of at least twelve months from
the date of the filing of these consolidated financial statements. The Company plans to raise the necessary additional capital through
one or a combination of public or private equity offerings, debt financings, and collaborations. The consolidated financial statements
do not include any adjustments that might result from the outcome of this uncertainty.
Notice from Nasdaq
On May 7, 2024, the Company received a letter
from the Nasdaq Listing Qualifications Staff of The Nasdaq Stock Market LLC (“Nasdaq”) stating that for the 30 consecutive
business day period between March 25, 2024 and May 6, 2024, the common stock of the Company had not maintained a minimum closing bid
price of $1.00 per share required for continued listing on The Nasdaq Capital Market pursuant to Nasdaq Listing Rule 5550(a)(2) (the
“Bid Price Rule”). Pursuant to Nasdaq Listing Rule 5810(c)(3)(A), the Company was provided an initial period of 180 calendar
days, or until November 4, 2024 (the “Compliance Period”), to regain compliance with the Bid Price Rule.
If the Company does not regain compliance
with the Bid Price Rule by November 4, 2024, the Company may be eligible for an additional 180-day period to regain compliance. To qualify,
the Company would be required to meet the continued listing requirement for market value of publicly held shares and all other initial
listing standards for The Nasdaq Capital Market, with the exception of the Bid Price Rule, and would need to provide written notice of
its intention to cure the bid price deficiency during the second compliance period, by effecting a reverse stock split, if necessary.
If the Company cannot regain compliance during
the Compliance Period or any subsequently granted compliance period, the common stock of the Company will be subject to delisting. At
that time, the Company may appeal the delisting determination to a Nasdaq hearings panel.
The notice from Nasdaq has no immediate effect
on the listing of the Company’s common stock. The Company is currently evaluating its options for regaining compliance. On December
21, 2023, the Company’s shareholders approved an amendment to its Certificate of Incorporation to provide for a reverse stock split
in a ratio ranging from one for two (1:2) to one for fifty (1:50) at the discretion of the Company’s Board of Directors.
Use of estimates
The preparation of the consolidated financial
statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect certain reported amounts and
disclosures. Accordingly, actual results could differ from those estimates. Significant estimates made by management include, but are
not limited to, realization of deferred tax assets, accrued liabilities, accrued commissions, incremental borrowing rate, obsolescence
of inventory and stock-based compensation.
Reverse Stock Split
On November 2, 2023, the Company effected
a 1-for-10 reverse stock split by filing an amendment to the Company’s Amended and Restated Certificate of Incorporation, as amended,
with the Delaware Secretary of State. The reverse stock split combined every ten shares of our common stock issued and outstanding immediately
prior to effecting the reverse stock split into one share of common stock. No fractional shares were issued in connection with the reverse
stock split. All historical share and per share amounts reflected throughout this document have been adjusted to reflect the reverse
stock split. The authorized number of shares and the par value per share of the Company’s common stock were not affected by the
reverse stock split.
Income Taxes
The Company accounts for income taxes utilizing
ASC 740, “Income Taxes”. ASC 740 requires the measurement of deferred tax assets for deductible temporary differences and
operating loss carry forwards, and of deferred tax liabilities for taxable temporary differences. Measurement of current and deferred
tax liabilities and assets is based on provisions of enacted tax law. The effects of future changes in tax laws or rates are not included
in the measurement. The Company recognizes the amount of taxes payable or refundable for the current year and recognizes deferred tax
liabilities and assets for the expected future tax consequences of events and transactions that have been recognized in the Company’s
financial statements or tax returns. The Company currently has substantial net operating loss carry forwards. The Company has recorded
a 100% valuation allowance against net deferred tax assets due to uncertainty of their ultimate realization. Valuation allowances are
established when necessary to reduce deferred tax assets to the amount expected to be realized.
Net loss per share
Basic net loss per share is based upon the
weighted average number of common shares outstanding. Diluted net loss per share is based on the assumption that all potential common
stock equivalents (convertible preferred stock, stock options, and warrants) are converted or exercised. The calculation of diluted net
loss per share excludes potential common stock equivalents if the effect is anti-dilutive. The Company’s weighted average common
shares outstanding for basic and diluted are the same because the effect of the potential common stock equivalents is anti-dilutive.
The Company had the following dilutive common
stock equivalents as of June 30, 2024 and 2023 which were excluded from the calculation because their effect was anti-dilutive:
| | |
June 30, 2024 | | |
June 30, 2023 | |
| Outstanding restricted stock units | |
| 136,690 | | |
| 93,156 | |
| Outstanding stock options | |
| 72,563 | | |
| 96,934 | |
| Outstanding warrants | |
| 2,388,068 | | |
| 2,009,600 | |
| Common shares convertible from preferred stock | |
| 2,625,016 | | |
| — | |
| Total | |
| 5,222,337 | | |
| 2,199,690 | |
Recent
Accounting Pronouncements Not Yet Adopted
In November 2023, the Financial Accounting
Standards Board (the “FASB”) issued Accounting Standards Update No. 2023-07, Segment Reporting (Topic 280): Improvements
to Reportable Segment Disclosures, which increases the disclosures about reportable segments including more detailed information
about a reportable segment’s expenses. This guidance will be effective for the Company for the fiscal year ending December 31,
2024 and the interim periods thereafter, with early adoption permitted. The guidance will have no effect on the Company’s results
of operations as the changes are disclosure related. The Company has elected not to early adopt.
In December 2023, the FASB issued Accounting
Standards Update 2023-09, “Income Taxes (Topic 740) - Improvements to Income Tax Disclosures”, which requires
additional tax disclosures about a reporting entity’s effective tax rate reconciliation as well as information on income taxes
paid. This guidance will be effective on a prospective basis, with the option to apply it retrospectively, for fiscal years beginning
after December 15, 2024. We are currently evaluating the impact of adopting this new accounting guidance.
3. Fixed Assets, Net
Fixed assets, net, consisted of the following:
| | |
June 30, 2024 | | |
December 31, 2023 | |
| Construction in progress | |
$ | 718 | | |
$ | 602 | |
| Catamaran tray sets | |
| 538 | | |
| 538 | |
| IT equipment | |
| 56 | | |
| 56 | |
| Leasehold improvements | |
| 15 | | |
| 15 | |
| Lab equipment | |
| 14 | | |
| 14 | |
| Office furniture | |
| 9 | | |
| 9 | |
| Fixed assets, gross | |
| 1,350 | | |
| 1,234 | |
| Less: accumulated depreciation | |
| (446 | ) | |
| (273 | ) |
| Fixed assets, net | |
$ | 904 | | |
$ | 961 | |
Construction in progress is made up of reusable
components that will become reusable Catamaran Tray Sets. Depreciation expense was approximately $86 and $35 for the three months ended
June 30, 2024 and 2023, respectively. Depreciation expense was approximately $173 and $60 for the six months ended June 30, 2024 and 2023,
respectively.
4. Accrued Expenses
Accrued expenses consisted of the following:
| | |
June 30, 2024 | | |
December 31, 2023 | |
| Accrued compensation | |
$ | 531 | | |
$ | 334 | |
| Other accrued expenses | |
| 401 | | |
| 474 | |
| Total accrued expenses | |
$ | 932 | | |
$ | 808 | |
5. Debt
Convertible notes payable
In November 2023, the Company entered into
Securities Purchase Agreements with certain investors (the “Investors”), pursuant to which the Company sold to the Investors
a total of $1,250,000 in secured notes (the “Convertible Notes”) and warrants to purchase 45,000 shares of the Company’s
common stock at an exercise price equal to $1.94 per share.
The Convertible Notes bear an interest rate
of 10% per annum with a default rate of 12% per annum and have a maturity date of November 21, 2024. All principal and accrued interest
is payable at maturity. At any time during the term of the Convertible Notes, the principal amount together with all accrued interest
thereon (the “Prepayment Amount”) may be paid in full, but not in part, by the Company. The Prepayment Amount may be paid
by the Company in cash or by the issuance to the Investors of shares of Series A Preferred Stock, if prior to such payment with Series
A Preferred Stock (i) certain stockholder proposals described in the Convertible Notes are approved by the Company’s stockholders;
and (ii) the Company has commitments from investors other than the Investors to purchase shares of Series A Preferred Stock with a stated
value of at least $3,750,000. The Convertible Notes are secured by a first priority security interest in all of the assets of the Company.
The warrants expire five years from the issuance date. The warrants contain a “cashless exercise” feature and contain anti-dilution
rights on subsequent issuances of equity or equity equivalents.
On February 20, 2024, the Investors agreed
to a complete prepayment of the Company’s obligations under the Convertible Notes, including accrued interest, in exchange for
84,729 shares of Series A Preferred Stock and warrants to purchase 157,094 shares of our common stock at $1.2705 per share and the Convertible
Notes were cancelled. See Note 7.
6. Leases
In June 2021, the Company entered into a facility
lease agreement for its company headquarters in Los Gatos, California. This non-cancellable operating lease expires in June 2026. Operating
lease costs for the facility lease were $73 and $73 for the three months ended June 30, 2024 and 2023, respectively, and were $146 and
$146 for the six months ended June 30, 2024 and 2023, respectively.
Supplemental balance sheet information related
to leases was as follows:
| | |
June 30, | | |
December 31, | |
| | |
2024 | | |
2023 | |
| Operating lease right-of-use assets | |
$ | 525 | | |
$ | 646 | |
| | |
| | | |
| | |
| Operating lease liability, current | |
$ | (271 | ) | |
$ | (256 | ) |
| Operating lease liability, noncurrent | |
| (290 | ) | |
| (428 | ) |
| Total operating lease liabilities | |
$ | (561 | ) | |
$ | (684 | ) |
Future maturities of operating lease liabilities
as of June 30, 2024 were as follows:
| 2024 | |
$ | 154 | |
| 2025 | |
| 310 | |
| 2026 | |
| 144 | |
| Total lease payments | |
| 608 | |
| Less: imputed interest | |
| (47 | ) |
| Present value of operating lease liabilities | |
$ | 561 | |
Other information:
| Cash paid for operating leases for the six months ended June 30, 2024 | | $ | 148 | |
| Cash paid for operating leases for the six months ended June 30, 2023 | | $ | 144 | |
| Remaining lease term - operating leases (in years) | | | 2.00 | |
| Average discount rate - operating leases | | | 8.0 | % |
7. Stockholders’ Equity
The Company’s current Amended and Restated
Certificate of Incorporation dated February 18, 2014 authorizes the issuance of 130,000,000 shares of common stock and 20,000,000 shares
of preferred stock, both with a par value of $0.001 per share. With respect to the preferred stock, 4,500,000 shares are designated Series
A Preferred Stock and 491,222 shares are designated Series B Preferred Stock.
At-the-Market Offering Program
On May 4, 2023, the Company entered into an Equity
Distribution Agreement to establish an at-the-market offering program, under which the Company may sell from time to time, at its option,
shares of its common stock having an aggregate gross sales price of $5.5 million. The Company is required to pay the Sales Agents a commission
of 3% of the gross proceeds from the sale of shares and has also agreed to provide the Sales Agents with customary indemnification rights.
During the six months ended June 30, 2024, 1,033,592 shares of the Company’s common stock were sold under the program at a weighted-average
price of $1.83 per share with aggregate proceeds, net of issuance costs, of $1,834. No shares were sold under the program during the three
months ended June 30, 2024. As of the date of this report, the Company may not sell additional shares under this program.
Equity Line of Credit
On July 24, 2023, the Company entered
into a purchase agreement (“Purchase Agreement”) with Lincoln Park Capital Fund, LLC (“Lincoln Park”), under
which, subject to specified terms and conditions, the Company may sell to Lincoln Park up to $10 million of shares of common stock from
time to time during the term of the Purchase Agreement. On September 22, 2023 (the “Commencement Date”) and on May 10, 2024,
the Company filed registration statements with the SEC covering the resale of shares of common stock issued to Lincoln Park under the
Purchase Agreement.
Beginning on the Commencement Date and for
a period of 24 months thereafter, under the terms and subject to the conditions of the Purchase Agreement, from time to time, at the
Company’s discretion, the Company has the right, but not the obligation, to sell to Lincoln Park, and Lincoln Park is obligated
to purchase, up to $10 million of shares of common stock, subject to certain limitations set forth in the Purchase Agreement. Specifically,
from time to time from and after the Commencement Date, the Company may, at its discretion, direct Lincoln Park to purchase on any single
business day on which the closing price of its common stock on The Nasdaq Capital Market (“Nasdaq”) is equal to or greater
than $1.50 up to 10,000 shares of common stock (a “Regular Purchase”); provided, that the Company may direct Lincoln Park
to purchase in a Regular Purchase (i) up to 12,500 shares of common stock, if the closing sale price of its common stock on Nasdaq on
such business day is at least $15.00 per share and (ii) up to 15,000 shares of common stock, if the closing sale price of its common
stock on Nasdaq on such business day is at least $25.00 per share. In no case, however, will Lincoln Park’s commitment with respect
to any single Regular Purchase exceed $500,000; provided, that the parties may mutually agree at any time to increase the maximum number
of shares of common stock the Company may direct Lincoln Park to purchase in any single Regular Purchase to up to 100,000 shares or any
number of shares that shall not exceed 4.99% of the then outstanding shares of common stock. The foregoing share amounts and per share
prices will be adjusted for any reorganization, recapitalization, non-cash dividend, stock split, reverse stock split or other similar
transaction occurring after the date of the Purchase Agreement with respect to our common stock. The purchase price per share for each
such Regular Purchase will be based on prevailing market prices of the Company’s common stock immediately preceding the time of
sale, as determined under the Purchase Agreement.
During the six months ended June 30, 2024, 89,847
shares of the Company’s common stock were sold under the program at a weighted-average price of $1.113 per share with aggregate
net proceeds of $96. No shares were sold under the program during the three months ended June 30, 2024.
Series A Preferred Stock
On February 20, 2024, the Company entered
into a Securities Purchase Agreement (the “Purchase Agreement”) with certain investors, pursuant to which the Company agreed
to sell, issue and deliver to these investors, in a private placement offering (the “Offering”), a total of 172,239 shares
of the Company’s Series A Preferred Stock and warrants (the “Series A Warrants”) to purchase 258,374 shares of common
stock, par value $0.001 per share, of the Company (“Common Stock”) at an exercise price equal to $1.2705 per share for an
aggregate offering price of $2,605,000.
Additionally, on February 20, 2024, the Investors
agreed to a complete prepayment of the Company’s obligations under the Convertible Notes, including accrued interest, in exchange
for 84,729 shares of Series A Preferred Stock and warrants to purchase 157,094 shares of our common stock at $1.2705 per share and the
Convertible Notes were cancelled. The Series A Warrants are immediately exercisable and expire five years from the date of issuance.
The Series A Preferred Stock is convertible,
at any time, at the option of the holder into shares of Common Stock. Each share of Series A Preferred Stock shall be convertible, at
any time after the date of issuance, at the option of the holder thereof (or, upon a Required Conversion (as defined below), at the option
of the Corporation), into that number of shares of Common Stock determined by dividing the Stated Value (as defined below) for such share
of Series A Preferred Stock by the Conversion Price (as defined below). “Stated Value” means for any share of Series A Preferred
Stock, an amount equal to the product of (x) $15.125 multiplied by (y) the sum of 1 plus the product of (A) 0.06 multiplied by (B) a
fraction equal to the number of days that such share of Series A Preferred Stock has been issued divided by 365. “Conversion Price”
means (i) for the shares of Series A Preferred Stock issued on the Closing Date, $1.5125 and (ii) for each share of Series A Preferred
Stock issued thereafter, an amount equal to the greater of (x) $1.5125 and the average of the VWAPs for the 10 Trading Days prior the
issuance date of such share of Series A Preferred Stock, in each case subject to adjustment as set forth herein. On any date that ten
out of the last 15 daily VWAPs of the Common Stock is 250% higher than the Conversion Price on such date, then the Company will have
the right to require 50% of the Preferred Stock to be converted into shares of Common Stock. Additionally, on and after the time on which
the Company has $2.25 million in revenues in any single financial quarter, the Company will have the right to require 50% of the Preferred
Stock to be converted into shares of Common Stock (a “Required Conversion”). No dividends are payable on the Series A Preferred
Stock. The Series A Preferred Stock will vote together with the Common Stock on all matters other than as required by law; provided however
that any additional shares underlying the Series A Preferred Stock as a result of the anti-dilution provision described below shall not
vote on an “as converted” basis and shall only vote when issued upon conversion. Notwithstanding the foregoing, the vote
of an individual holder of Series A Preferred Stock (and underlying Common Stock) shall be capped at 9.99% (or 4.99% if selected by the
holder).
The Conversion Price is subject to anti-dilution
adjustment as the result of any subdivision, combination of shares or recapitalization, stock dividends, stock splits and similar transactions
affecting the Common Stock. In addition, the Series A Preferred Stock will have weighted average anti-dilution protection providing for
adjustment of the Conversion Price in the event of issuance of, or commitments to issue, Common Stock for less than the Conversion Price
then in effect immediately prior to such issue or sale (a “Dilutive Issuance”), subject to customary exceptions; provided
however the anti-dilution for Dilutive Issuances shall not be operative until the stockholders of the Company have approved the terms
of the Series A Preferred Stock. Upon any liquidation or winding up of the Company (a “Liquidation”), the holders of Series
A Preferred Stock will be entitled to receive in preference to any other class or series of the Company’s equity securities the
greater of (i) the Stated Value plus accrued and unpaid dividends and (ii) what would be paid if the Series A Preferred Stock plus accrued
and unpaid dividends had been converted into Common Stock. A consolidation or merger of the Company or sale or transfer of all or substantially
all of its assets, or any transaction which results in the stockholders of the Company owning less than 50% of the equity or voting power
of the surviving entity (excluding the issuance of Common Stock in any financing transaction unless more than 50% of the Company’s
shares are issued to one stockholder or a number of stockholders who act as a one group) shall be deemed a Liquidation (a “Deemed
Liquidation”) with respect to the shares of Series A Preferred Stock of any holder who opts to have such occurrence treated as
a Deemed Liquidation; provided that if the liquidation preference payable on a Deemed Liquidation is less than 110% of the stated value
of the Series A Preferred Stock, the dividend rate on any accrued and unpaid dividends payable with respect to such Deemed Liquidation
will increase to 10%. All liquidation preferences payable in respect of a Deemed Liquidation will be payable in shares of Common Stock
based on the closing price of the Common Stock on the date of such Deemed Liquidation. Consent of the majority of the holders will be
required to (i) amend the Certificate of Incorporation or Bylaws of the Company so as to adversely alter the rights, preferences, privileges
of the Series A Preferred Stock, (ii) create any new class of shares pari passu or senior to the Series A Preferred Stock or increase
or decrease the number of authorized shares of Common Stock or preferred stock, (iii) pay or declare any dividend on Common Stock or
other junior securities, or incur indebtedness in any single transaction in excess of $1 million or (iv) redeem, purchase or otherwise
acquire any share or shares of preferred stock or Common Stock (other than (a) the repurchase of shares of Common Stock pursuant to a
written benefit plan or employment or consulting agreement, or (b) the repurchase of any equity securities in connection with the Company’s
right of first offer with respect to those securities contained in any written agreement with the Company).
Voting rights
The holders of vested shares of common stock
are entitled to vote on any matter submitted to a vote of the stockholders and each such holder is entitled to one vote per share of
common stock held. The holders of Series A and Series B Preferred Stock are entitled to vote together with the common stock as a single
class on any matter submitted to a vote of the stockholders. Holders of Series A and Series B Preferred Stock are entitled to the number
of votes equal to the number of common stock issuable upon conversion of their respective Series A and Series B Preferred Stock at the
time such shares are voted. The holders of a majority of the preferred stock had additional voting rights as specified in the Company’s
Amended and Restated Certificate of Incorporation, as amended.
Equity awards
In 2012, the Board of Directors of the Company
(the “Board”) approved the Tenon Medical, Inc. 2012 Equity Incentive Plan (the “2012 Plan”). The 2012 Plan provides
for the issuance of common stock options, appreciation rights, and other awards to employees, directors, and consultants. Options issued
under the 2012 Plan generally vest over a period of two to four years and have a 10-year expiration date. In April 2021, the Board increased
the number of shares of common stock reserved for issuance under the 2012 Plan to 662,516. In July 2021, the Board increased the number
of shares of common stock reserved for issuance under the 2012 Plan to 737,516. In August 2021, the Board increased the number of shares
of common stock reserved for issuance under the 2012 Plan from 737,516 shares to 799,266 shares and approved the form of a 2022 Equity
Incentive Plan.
On January 10, 2022 and February 2, 2022,
the Board and stockholders, respectively, of the Company approved the Tenon Medical, Inc. 2022 Equity Incentive Plan (the “2022
Plan”), which was effective on April 25, 2022. The number of shares of common stock that may be subject to awards and sold under
the 2022 Plan is equal to 1,600,000. Automatic annual increases in number of shares available for issuance under the 2022 Plan is equal
to the least of (a) 1,100,000 shares, (b) 4% of the total number of shares of all classes of common stock outstanding on the last day
of the immediately preceding fiscal year, or (c) such number determined by the 2022 Plan administrator no later than the last day of
the immediately preceding fiscal year. Annual increases will continue until the tenth anniversary of the earlier of the Board or stockholder
approval of the 2022 Plan, which is January 10, 2032. Upon the effective date of the 2022 Plan, the Board terminated the 2012 Plan such
that no new equity awards will be issued by the 2012 Plan.
Option Exchange
On April 8, 2024, the Company issued an offer
to holders of outstanding stock options to purchase an aggregate of 90,987 shares of the Company’s common stock to exchange their
options for a lesser number of new restricted stock units (“RSUs”) to be granted under the 2022 Plan upon the terms and subject
to the conditions set forth in the Offer to Exchange Certain Outstanding Stock Options for Restricted Stock Units (the “Offer to
Exchange”). The Offer to Exchange expired on May 6, 2024. A total of approximately 27 eligible participants participated in the
exchange. The Company accepted for exchange options to purchase an aggregate of approximately 83,391 shares of common stock of the Company.
All surrendered options were cancelled effective as of the expiration of the Option Exchange, and immediately thereafter, in exchange
therefor, the Company granted a total of approximately 41,698 new RSUs under the 2022 Plan.
A summary of the Company’s stock option
and restricted stock unit activity under its plans is as follows:
| | |
Number of Shares
Subject to
Outstanding Stock Options | | |
Weighted
Average
Exercise
Price per
Share | | |
Number of Outstanding Restricted Stock Units | | |
Weighted Average Grant Date Fair Value per Unit | |
| Outstanding at December 31, 2023 | |
| 102,089 | | |
$ | 42.54 | | |
| 76,916 | | |
$ | 69.50 | |
| Granted | |
| 66,967 | | |
$ | 0.87 | | |
| 119,351 | | |
$ | 1.00 | |
| Released | |
| — | | |
| — | | |
| (57,077 | ) | |
$ | 32.05 | |
| Cancelled or forfeited | |
| (96,493 | ) | |
$ | 43.27 | | |
| (2,500 | ) | |
$ | 2.91 | |
| Outstanding at June 30, 2024 | |
| 72,563 | | |
$ | 3.12 | | |
| 136,690 | | |
$ | 26,55 | |
The following table sets forth stock-based compensation
expense recognized for the three and six months ended June 30, 2024 and 2023:
| | |
Three months ended
June 30, | | |
Six months ended
June 30, | |
| | |
2024 | | |
2023 | | |
2024 | | |
2023 | |
| Research and development | |
$ | 358 | | |
$ | 378 | | |
$ | 725 | | |
$ | 750 | |
| Sales and marketing | |
| 34 | | |
| 58 | | |
| 77 | | |
| 116 | |
| General, and administrative | |
| 642 | | |
| 618 | | |
| 1,250 | | |
| 1,228 | |
| Total stock-based compensation expense | |
$ | 1,034 | | |
$ | 1,054 | | |
$ | 2,052 | | |
$ | 2,094 | |
At June 30, 2024, there were 54,173 shares available
for issuance under the 2022 Plan.
Warrants
In April 2022, in association with the Company’s
initial public offering, the Company granted to The Benchmark Company, LLC and Valuable Capital Limited warrants to purchase a total
of 9,600 shares of the Company’s common stock. The warrants were immediately exercisable at an exercise price of $50.00 per share
and expire on the fifth anniversary of the commencement of sales under the IPO. The fair value of the warrants on the grant date was
$27.50 per warrant, which was calculated using a Black-Scholes option valuation model with an expected term of 5.00 years, expected volatility
of 62.55%, dividend yield of 0%, and risk-free interest rate of 2.92%. The Company recorded the fair value of these warrants of approximately
$264 as an issuance cost to additional paid-in capital in 2022.
In June 2023, in connection with a registered
offering of stock, the Company issued warrants to purchase a total of 2,000,000 shares of the Company’s common stock (the “Offering
Warrants”). The Offering Warrants were exercisable upon issuance and will expire five years from the date of issuance. Per the
terms of the Offering Warrants, the exercise price of the Offering Warrants reset on July 16, 2023, to $3.146 per share. The fair value
of the Offering Warrants on the grant date was approximately $3,164, or $1.58 per warrant, which was calculated using a Monte-Carlo simulation
to estimate the final exercise price, which is considered a Level 3 fair value measurement, using as inputs; the starting value of $3.00
per share, the Company’s VWAP on June 16; an assumed daily distribution of returns; a mean daily return of 5.18%; a short-term
annual volatility of 100% and a standard deviation of 6.3%. The model used Black-Scholes to then calculate the estimated fair value of
the Offering Warrants, using an estimated time to maturity of 4.9 years, a risk-free interest rate of 3.99% and a long-term volatility
of 60%.
In November 2023, in connection with the issuance
of the Convertible Notes, the Company issued warrants to purchase a total of 45,000 shares of the Company’s common stock at an
exercise price equal to $1.94 per share. The warrants expire five years from the issuance date. The fair value of the warrants on the
grant date was $1.29 per warrant, which was calculated using a Black-Scholes option valuation model with an expected term of 5.00 years,
expected volatility of 68.89%, dividend yield of 0%, and risk-free interest rate of 4.41%. The Company recorded the fair value of these
warrants of approximately $58 as an issuance cost to additional paid-in capital in 2023.
On February 20, 2024, in connection with the
Purchase Agreement, the Company issued the Series A Warrants to purchase a total of 415,468 shares of the Company’s common stock
at an exercise price equal to $1.2705 per share. The Series A Warrants are immediately exercisable and expire five years from the date
of issuance. The fair value of the Series A Warrants on the grant date was $0.61 per warrant, which was calculated using a Black-Scholes
option valuation model with an expected term of 5.00 years, expected volatility of 68.24%, dividend yield of 0%, and risk-free interest
rate of 4.3%. The Company recorded the fair value of these warrants of approximately $254 to additional paid-in capital in 2024.
8. Commitments and Contingencies
Sales Representative Agreement
In April 2020, the Company entered into an
Exclusive Sales Representative Agreement, under which the counterparty to the agreement (the “Representative”) received exclusive
rights to market, promote, and distribute The Catamaran System in the United States and Puerto Rico. The agreement is for an initial
period of five years, and automatically renews for an additional five years unless written notice is given by either party prior to April
27, 2023. The agreement provides for a bonus to be paid to the Representative upon an acquisition or IPO. In May 2021, the Company entered
into an Amended and Restated Exclusive Sales Representative Agreement (the “Restated Sales Agreement”). In connection with
the amended agreement, the Company paid $500 cash and issued 53,757 shares of common stock to the Representative, for which the Company
recorded a combined total of approximately $880 as sales and marketing expense. In addition, the Representative received anti-dilution
protections to maintain ownership of 3.0% of the fully diluted equity of the Company through the date of an initial public offering.
In October 2021, the Company issued 4,445 shares of common stock with a fair value of approximately $333 to the Representative in accordance
with the anti-dilution provision. In April 2022, the Company issued 31,235 shares of common stock to the Representative in accordance
with the anti-dilution provision, fully satisfying the Company’s obligations.
The Restated Sales Agreement restructured
the calculation of the bonus paid to the Representative upon an acquisition, removed the bonus payable upon an IPO, and allows the Company
to terminate the Restated Sales Agreement as long as the bonus paid to the Representative is at least $6,000.
On October 6, 2022, the Company entered into the
Terminating Amended and Restated Exclusive Sales Representative Agreement (the “Termination Agreement”) with the Representative,
which terminated the Restated Sales Agreement. In accordance with the Termination Agreement, (i) the Company paid the Representative $1,000
in cash; and (ii) the Company agreed to pay the Representative (a) $85 per month during the six months after the date of the Termination
Agreement in return for efforts by the Representative to transition operations to the Company, (b) 20% of net sales of the product sold
in the United States and Puerto Rico until December 31, 2023 and (c) after December 31, 2023, 10% of net sales until such time as the
aggregate amount paid to the Representative under this clause (c) and clause (b) above equal $3,600. In the event of an acquisition of
the Company, the Company will pay the Representative $3,600 less previous amounts paid pursuant to clause (b) and clause (c) above. The
Company recorded a charge of $1,000 for the payment to the Representative in the fourth quarter of 2022 and expensed the $85 per month
charges as incurred over the six-month period. For payments under clause (b) and clause (c) above, the Company estimated the fair value
of the liability using level 3 hierarchy inputs based on a Monte Carlo simulation of future revenues with a 25% quarterly estimated standard
deviation of growth rates and a 10% probability of dissolution, discounted at an estimated discount rate of 15.4%. Based on the Company’s
fair value analysis, a total of $2,611 was charged to sales and marketing expense in the consolidated statements of operations and comprehensive
loss and recorded as accrued commissions in the consolidated balance sheets. A reconciliation of the liability under clause (b) and clause
(c) for the six months ended June 31, 2024 is as follows:
| Balance at January 1, 2024 | |
$ | 2,377 | |
| Amounts paid during 2024 | |
| (252 | ) |
| Accretion | |
| 185 | |
| Balance at June 30, 2024 | |
$ | 2,310 | |
Per the terms of the Termination Agreement,
the Company ultimately expects to expense $3,600 under clause (b) and clause (c).
Simultaneously with the execution of the Termination
Agreement, the Company entered into a Consulting Agreement dated October 6, 2022, with the Representative (the “Consulting Agreement”).
Under the terms and conditions of the Consulting Agreement, the Representative is tasked with organizing, recruiting, training, and coordinating
the Company’s Clinical Specialist program, Physician Education program and Sales Education program as more specifically described
in the Consulting Agreement.
The term of the Consulting Agreement was from
October 6, 2022, until October 5, 2023, when it terminated in accordance with the terms of the Consulting Agreement. In consideration
for the services to be provided, the Company paid the Representative a base consulting fee of $700 per year, payable in monthly instalments,
along with additional compensation of $62.5 per quarter, if certain sales targets were met, for four quarters; along with any travel
and related out-of-pocket expenses incurred by the Representative in connection with the performance of the services.
Litigation
In the normal course of business, the Company
may possibly be named as a defendant in various lawsuits.
9. Concentrations of Risk
Credit risk
Financial instruments that potentially subject
the Company to concentrations of credit risk consist principally of cash and cash equivalents.
The Company maintains cash balances at financial
institutions located in California. Accounts at the U.S. financial institutions are secured by the Federal Deposit Insurance Corporation.
At times, balances may exceed federally insured limits. The Company has not experienced any losses in such accounts. Management believes
that the Company is not exposed to any significant credit risk with respect to its cash and cash equivalents.
The Company grants unsecured credit to its
customers based on an evaluation of the customer’s financial condition and a cash deposit is generally not required. Management
believes its credit policies do not result in significant adverse risk and historically has not experienced significant credit-related
losses.
Up to [*] shares of Common Stock
Common Warrants to Purchase up to [*] shares
of Common Stock
Pre-funded Warrants to Purchase up to [*] shares
of Common Stock
Up to [*] shares of Common Stock underlying
the Common Warrants
Up to [*] shares of Common Stock underlying
the Pre-funded Warrants
Tenon Medical,
Inc.
PROSPECTUS
Part II
INFORMATION NOT REQUIRED
IN PROSPECTUS
Item 13. Other Expenses
of Issuance and Distribution.
The
following table indicates the expenses to be incurred in connection with the offering described in this registration statement, other
than placement agent fees and commissions, all of which will be paid by us. All amounts are estimated except the Securities and Exchange
Commission registration fee and the Financial Industry Regulatory Authority, Inc., or FINRA filing.
| | |
Amount | |
| Securities and Exchange Commission registration fee | |
$ | | |
| FINRA filing fee | |
$ | | |
| Accountants’ fees and expenses | |
$ | | |
| Legal fees and expenses | |
$ | | |
| Printing and engraving expenses | |
$ | | |
| Miscellaneous | |
$ | | |
| Total expenses | |
$ | | |
Item 14. Indemnification
of Directors and Officers.
Section
102 of the General Company Law of the State of Delaware (“DGCL”) permits a Company to eliminate the personal liability of
directors of a Company to the Company or its stockholders for monetary damages for a breach of fiduciary duty as a director, except where
the director breached his duty of loyalty, failed to act in good faith, engaged in intentional misconduct or knowingly violated a law,
authorized the payment of a dividend or approved a stock repurchase in violation of Delaware corporate law or obtained an improper personal
benefit. Our charter, as amended provides that no director of the Company shall be personally liable to it or its stockholders for monetary
damages for any breach of fiduciary duty as a director, notwithstanding any provision of law imposing such liability, except to the extent
that the DGCL prohibits the elimination or limitation of liability of directors for breaches of fiduciary duty.
Section
145 of the DGCL provides that a Company has the power to indemnify a director, officer, employee, or agent of the Company, or a person
serving at the request of the Company for another Company, partnership, joint venture, trust or other enterprise in related capacities
against expenses (including attorneys’ fees), judgments, fines and amounts paid in settlement actually and reasonably incurred by
the person in connection with an action, suit or proceeding to which he was or is a party or is threatened to be made a party to any threatened,
ending or completed action, suit or proceeding by reason of such position, if such person acted in good faith and in a manner he reasonably
believed to be in or not opposed to the best interests of the Company, and, in any criminal action or proceeding, had no reasonable cause
to believe his conduct was unlawful, except that, in the case of actions brought by or in the right of the Company, no indemnification
shall be made with respect to any claim, issue or matter as to which such person shall have been adjudged to be liable to the Company
unless and only to the extent that the Court of Chancery or other adjudicating court determines that, despite the adjudication of liability
but in view of all of the circumstances of the case, such person is fairly and reasonably entitled to indemnity for such expenses which
the Court of Chancery or such other court shall deem proper.
Our charter, as amended provides
that we will indemnify to the fullest extent permitted from time to time by the DGCL or any other applicable laws as presently or hereafter
in effect, any person who was or is a party or is threatened to be made a party to any threatened, pending or completed action, suit or
proceeding, whether civil, criminal, administrative or investigative, including, without limitation, an action by or in the right of the
Company, by reason of his acting as a director or officer of the Company or any of its subsidiaries (and the Company, in the discretion
of the Board of Directors, may so indemnify a person by reason of the fact that he is or was an employee or agent of the Company or any
of its subsidiaries or is or was serving at the request of the Company in any other capacity for or on behalf of the Company) against
any liability or expense actually and reasonably incurred by such person in respect thereof; provided,
however, the Company shall be required to indemnify an officer or director in connection with an action, suit or proceeding
(or part thereof) initiated by such person only if (i) such action, suit or proceeding (or part thereof) was authorized by the Board of
Directors and (ii) the indemnification does not relate to any liability arising under Section 16(b) of the Exchange Act, as amended, or
any rules or regulations promulgated thereunder. Such indemnification is not exclusive of any other right to indemnification provided
by law or otherwise.
If a claim is not paid in
full by the Company, the claimant may at any time thereafter bring suit against the Company to recover the unpaid amount of the claim
and, if successful in whole or in part, the claimant shall be entitled to be paid also the expense of prosecuting such claim. It shall
be a defense to any such action (other than an action brought to enforce a claim for expenses incurred in defending any proceeding in
advance of its final disposition where any undertaking required by the By-laws of the Company has been tendered to the Company) that the
claimant has not met the standards of conduct which make it permissible under the DGCL for the Company to indemnify the claimant for the
amount claimed, but the burden of proving such defense shall be on the Company. Neither the failure of the Company (including its Board
of Directors, legal counsel, or its stockholders) to have made a determination prior to the commencement of such action that indemnification
of the claimant is proper in the circumstances because he or she has met the applicable standard of conduct set forth in the DGCL, nor
an actual determination by the Company (including its Board of Directors, legal counsel, or its stockholders) that the claimant has not
met such applicable standard of conduct, shall be a defense to the action or create a presumption that the claimant has not met the applicable
standard of conduct. Indemnification shall include payment by the Company of expenses in defending an action or proceeding in advance
of the final disposition of such action or proceeding upon receipt of an undertaking by the person indemnified to repay such payment if
it is ultimately determined that such person is not entitled to indemnification.
In
any underwriting agreement we enter into in connection with the sale of common stock being registered hereby, the underwriters will agree
to indemnify, under certain conditions, us, our directors, our officers and persons who control us within the meaning of the Securities
Act of 1933, as amended, or the Securities Act, against certain liabilities.
Item 15. Recent Sales
of Unregistered Securities.
Set forth below is information regarding shares
of capital stock issued by us within the last three years which was not registered under the Securities Act of 1933, as amended.
(a) Issuance of Capital
Stock.
Common Stock and Preferred Stock
On August 10, 2021, the
Company issued an aggregate of 6,175 shares of restricted common stock to several individuals under the Tenon Medical, Inc. 2012 Equity
Incentive Plan.
During October 2021, the Company issued 4,445
shares of common stock to SpineSource, Inc. pursuant to an anti-dilution provision. The shares of common stock had a value of $74.90 per
share.
On October 28, 2021, the Company issued 255,077
shares of Series A preferred stock to Zuhlke Ventures AG pursuant to an Exchange Agreement based on a deemed value of $0.98 per share.
In April of 2022, we
issued 31,236 shares of common stock to SpineSource, Inc. The shares of common stock had a value of $50 per share.
In February 2024, we
issued a total of 256,968 shares of Series A Preferred Stock to investors.
In July of 2023, we completed a private placement
to Lincoln Park Capital Fund, LLC (“Lincoln Park”) pursuant to which we have the right to sell to Lincoln Park up to $10.0
million in shares of common stock, subject to certain limitations, from time to time over the 24-month period commencing on the date that
a registration statement covering the resale of the shares is declared effective by the SEC. We issued 98,909 Commitment Shares to Lincoln
Park as consideration for its commitment to purchase our shares under the Purchase Agreement. In the Purchase Agreement, Lincoln Park
represented to us, among other things, that it was an “accredited investor” (as such term is defined in Rule 501(a) of Regulation
D under the Securities Act). The securities were and will be sold by us under the Purchase Agreement in reliance upon an exemption from
the registration requirements under the Securities Act afforded by Section 4(a)(2) of the Securities Act.
In February 2024, we
issued 89,847 shares to Lincoln Park pursuant to a purchase facility with Lincoln Park.
In July 2024, we issued
170,940 shares to Lincoln Park pursuant to a purchase facility with Lincoln Park.
The issuance of the capital stock listed above
was deemed exempt from registration under Section 4(a)(2) of the Securities Act or Regulation D promulgated thereunder in that the issuance
of securities were made to an accredited investor and did not involve a public offering. The recipient of such securities represented
its intention to acquire the securities for investment purposes only and not with a view to or for sale in connection with any distribution
thereof.
(b) Option Grants.
On August 10, 2021 the Company issued a total
of 2,100 options to various individuals at an exercise price of $70.60 per share. The options are subject to equal monthly vesting over
a two or three-year period.
On October 8, 2021 the Company issued a total
of 2,200 options to various individuals at an exercise price of $75 per share. The options are subject to equal monthly vesting over a
two or three-year period.
The options described above were deemed exempt
from registration in reliance on Section 4(a)(2) of the Securities Act or Regulation D promulgated thereunder in that the issuance of
securities were made to an accredited investor and did not involve a public offering. The recipients of such securities represented its
intention to acquire the securities for investment purposes only and not with a view to or for sale in connection with any distribution
thereof.
(c) Warrants.
In April 2022, in connection with the Company’s
initial public offering, the Company granted the Underwriters warrants to purchase a total of 9,600 shares of the Company’s common
stock. The warrants are immediately exercisable at an exercise price of $50 per share and expire on the fifth anniversary of the commencement
of sales under the IPO.
In November 2023, we issued warrants to purchase
45,000 shares of our common stock at an exercise price equal to $1.94 per share to investors.
In February 2024, we issued warrants to purchase
415,468 shares of our common stock at an exercise price equal to $1.2705 per share to investors.
The warrant described above were deemed exempt
from registration in reliance on Section 4(a)(2) of the Securities Act or Regulation D promulgated thereunder in that the issuance of
securities were made to an accredited investor and did not involve a public offering. The recipients of such securities represented its
intention to acquire the securities for investment purposes only and not with a view to or for sale in connection with any distribution
thereof.
(d) Issuance of Notes.
None
Item 16. Exhibits
and Financial Statement Schedules.
(a) Exhibits:
EXHIBIT INDEX
| Exhibit No. |
|
Description |
| 1.1# |
|
Form of Placement Agent Agreement |
| 3.1** |
|
Second Amended and Restated Certificate of Incorporation of the Registrant. |
| 3.2* |
|
Bylaws of The Registrant. |
| 4.1# |
|
Form of Pre-Funded Warrant |
| 4.2# |
|
Form of Common Warrant |
| 5.1# |
|
Opinion of Counsel to Registrant. |
10.1*## |
|
Employment Agreement dated June 1, 2021 between Steven M. Foster and the Registrant. |
10.2*## |
|
Employment Agreement dated June 1, 2021 between Richard Ginn and the Registrant. |
10.3*## |
|
Consulting Agreement dated May 7, 2021 by and between Richard Ferrari and the Registrant. |
| 10.4** |
|
Tenon Medical 2022 Equity Incentive Plan. |
| 10.5*** |
|
Purchase Agreement dated as of July 24, 2023, by and between the Registrant and Lincoln Park Capital Fund, LLC. |
| 10.6*** |
|
Registration Rights Agreement dated as of July 24, 2023, by and between the Registrant and Lincoln Park Capital Fund, LLC. |
| 10.7**** |
|
Form of Securities Purchase Agreement entered into between the Registrant and investors in the February 2024 private placement. |
| 10.8**** |
|
Form of Warrant issued in the February 2024 private placement issued by the Registrant. |
| 10.9***** |
|
Form of Securities Purchase Agreement entered into between the Registrant and investors in the November 2023 private placement. |
| 10.10***** |
|
Form of Note issued by the Registrant in the November 2023 private placement. |
| 10.11***** |
|
Form of Warrant issued by the Registrant in the November 2023 private placement. |
| 10.12# |
|
Form of Warrant Agent Agreement |
| 10.13# |
|
Form of Securities Purchase Agreement |
| 16.1# |
|
Letter from Armanino, LLP dated September 11, 2023, regarding change in accountant |
| 21.1* |
|
List of Subsidiaries of the Registrant. |
| 23.1 |
|
Consent of Haskell & White LLP, dated August 13, 2024. |
| 23.2 |
|
Consent of Armanino, LLP, dated August 13, 2024. |
| 23.3# |
|
Consent of Counsel to Registrant (included in Exhibit 5.1). |
| 24.1 |
|
Power of Attorney (included on the signature page hereto) |
| 107 |
|
Filing Fee Table. |
| * |
Incorporated by reference to the Registrant’s Registration Statement No. 333-260931, filed on November 10, 2021. |
| ** |
Incorporated by reference to the Registrant’s Registration Statement No. 333-271648, originally filed on May 4, 2023. |
| *** |
Incorporated by reference to the Registrant’s Current Report on Form 8-K originally filed on July 28, 2023. |
| **** |
Incorporated by reference to the Registrant’s Current Report on Form 8-K originally filed on February 22, 2024. |
| ***** |
Incorporated by reference to the Registrant’s Current Report on Form 8-K originally filed on November 28, 2023. |
| |
# |
To be filed by amendment. |
| |
|
|
| |
## |
Denotes management compensation plan, agreement or arrangement. |
(b) Financial Statement
Schedules: All schedules are omitted because the required information is inapplicable or the information is presented in the financial
statements and the related notes.
Item 17. Undertakings.
The undersigned registrant hereby undertakes:
The undersigned registrant
hereby undertakes:
(1) To file, during any period
in which offers or sales are being made, a post-effective amendment to this registration statement:
(i) To include any prospectus
required by section 10(a)(3) of the Securities Act of 1933, as amended (the “Securities Act”);
(ii) To reflect in the prospectus
any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof)
which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding
the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed
that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the
form of prospectus filed with the Securities and Exchange Commission pursuant to Rule 424(b) if, in the aggregate, the changes in volume
and price represent no more than a 20 percent change in the maximum aggregate offering price set forth in the “Calculation of Registration
Fee” table in the effective registration statement; and
(iii) To include any material
information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to
such information in the registration statement; provided, however, that paragraphs (1)(i), (1)(ii) and (1)(iii) above do not apply if
the information required to be included in a post-effective amendment by those paragraphs is contained in reports filed with or furnished
to the Securities and Exchange Commission by the registrant pursuant to Section 13 or Section 15(d) of the Securities Exchange Act of
1934 that are incorporated by reference in the registration statement, or is contained in a form of prospectus filed pursuant to Rule
424(b) that is part of the registration statement.
(2) That, for the purpose
of determining any liability under the Securities Act, each such post-effective amendment shall be deemed to be a new registration statement
relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide
offering thereof.
(3) To remove from registration
by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering.
(4) That, for the purpose
of determining liability under the Securities Act to any purchaser:
(A) Each prospectus filed
by the registrant pursuant to Rule 424(b)(3) shall be deemed to be part of the registration statement as of the date the filed prospectus
was deemed part of and included in the registration statement; and
(B) Each prospectus filed
pursuant to Rule 424(b) as part of a registration statement relating to an offering, other than registration statements relying on Rule
430B or other than prospectuses filed in reliance on Rule 430A, shall be deemed to be part of and included in the registration statement
as of the date it is first used after effectiveness. Provided, however, that no statement made in a registration statement or prospectus
that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration
statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such
first use, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration
statement or made in any such document immediately prior to such date of first use.
(5) That for the purpose of
determining liability of the registrant under the Securities Act to any purchaser in the initial distribution of securities, the undersigned
registrant undertakes that in a primary offering of securities of the undersigned registrant pursuant to this registration statement,
regardless of the underwriting method used to sell the securities to the purchaser, if the securities are offered or sold to such purchaser
by means of any of the following communications, the undersigned registrant will be a seller to the purchaser and will be considered to
offer or sell such securities to such purchaser:
(i) Any preliminary prospectus
or prospectus of the undersigned registrant relating to the offering required to be filed pursuant to Rule 424;
(ii) Any free writing prospectus
relating to the offering prepared by or on behalf of the undersigned registrant or used or referred to by the undersigned registrant;
(iii) The portion of any other
free writing prospectus relating to the offering containing material information about the undersigned registrant or its securities provided
by or on behalf of the undersigned registrant; and
(iv) Any other communication
that is an offer in the offering made by the undersigned registrant to the purchaser.
Insofar as indemnification
for liabilities arising under the Securities Act may be permitted to directors, officers and controlling persons of the registrant pursuant
to any charter provision, by law or otherwise, the registrant has been advised that in the opinion of the Securities and Exchange Commission
such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable. In the event that a
claim for indemnification against such liabilities (other than payment by the registrant of expenses incurred or paid by a director, officer
or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer
or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the
matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification
by it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue.
The undersigned registrant hereby undertakes that:
(1) For purposes of determining
any liability under the Securities Act, the information omitted from the form of prospectus filed as part of this registration statement
in reliance upon Rule 430A and contained in a form of prospectus filed by the registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under
the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective.
(2) For the purpose of determining
any liability under the Securities Act, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration
statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial
bona fide offering thereof.
SIGNATURES
Pursuant to the requirements of the Securities
Act of 1933, the registrant has duly caused this Registration Statement to be signed on its behalf by the undersigned, thereunto duly
authorized in the City of New York, State of New York, on August 14, 2024.
| |
TENON MEDICAL, INC. |
| |
|
| |
By: |
/s/
Steven M. Foster |
| |
|
Steven M. Foster |
| |
|
Chief Executive Officer and President
(Principal Executive Officer) |
POWER
OF ATTORNEY
KNOW ALL PERSONS BY THESE
PRESENTS, that each person whose signature appears below constitutes and appoints Steven M. Foster and Jay Hanson, or either of them,
as his true and lawful attorneys-in-fact and agents, with full powers of substitution and resubstitution, for him and in his name, place
and stead, in any and all capacities, to sign any and all amendments to this Registration Statement (including post-effective amendments
and any related registration statements filed pursuant to Rule 462 and otherwise), and to file the same with all exhibits thereto, and
other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents
and full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith,
as fully for all intents and purposes as he might or could do in person, hereby ratifying and confirming that said attorney-in-fact and
agent, or any substitute or resubstitute, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities
Act of 1933, this Registration Statement has been signed by the following persons in the capacities and on the dates indicated.
| Name |
|
Position |
|
Date |
| |
|
|
|
|
| /s/ Steven M. Foster |
|
Chief Executive Officer and President, Director |
|
August 14, 2024 |
| Steven M. Foster |
|
(Principal Executive Officer) |
|
|
| |
|
|
|
|
| /s/ Richard Ginn |
|
Chief Technology Officer and Director |
|
August 14, 2024 |
| Richard Ginn |
|
|
|
|
| |
|
|
|
|
| /s/ Jay Hanson |
|
Director of SEC Reporting and Compliance |
|
August 14, 2024 |
| Jay Hanson |
|
(Principal Financial and Accounting Officer) |
|
|
| |
|
|
|
|
| /s/ Richard Ferrari |
|
Director |
|
August 14, 2024 |
| Richard Ferrari |
|
|
|
|
| |
|
|
|
|
| /s/ Ivan Howard |
|
Director |
|
August 14, 2024 |
| Ivan Howard |
|
|
|
|
| |
|
|
|
|
| /s/ Kristine Jacques |
|
Director |
|
August 14, 2024 |
| Kristine Jacques |
|
|
|
|
| |
|
|
|
|
| /s/ Robert K. Weigle |
|
Director |
|
August 14, 2024 |
| Robert K. Weigle |
|
|
|
|
| |
|
|
|
|
| /s/ Stephen H. Hochschuler, M.D. |
|
Director |
|
August 14, 2024 |
| Stephen H. Hochschuler, M.D. |
|
|
|
|
II-7
23.62
8.59
1814
801
1.02
2.24
3.28
7.50
1215
1305
3301
3749
2655
false
0001560293
0001560293
2024-01-01
2024-06-30
0001560293
2023-12-31
0001560293
2022-12-31
0001560293
2024-06-30
0001560293
us-gaap:SeriesAPreferredStockMember
2024-06-30
0001560293
us-gaap:SeriesAPreferredStockMember
2023-12-31
0001560293
2023-01-01
2023-12-31
0001560293
2022-01-01
2022-12-31
0001560293
2024-04-01
2024-06-30
0001560293
2023-04-01
2023-06-30
0001560293
2023-01-01
2023-06-30
0001560293
tnon:SeriesAConvertiblePreferredStockMember
us-gaap:PreferredStockMember
2021-12-31
0001560293
tnon:SeriesBConvertiblePreferredStockMember
us-gaap:PreferredStockMember
2021-12-31
0001560293
us-gaap:CommonStockMember
2021-12-31
0001560293
us-gaap:AdditionalPaidInCapitalMember
2021-12-31
0001560293
us-gaap:RetainedEarningsMember
2021-12-31
0001560293
us-gaap:AccumulatedOtherComprehensiveIncomeMember
2021-12-31
0001560293
2021-12-31
0001560293
tnon:SeriesAConvertiblePreferredStockMember
us-gaap:PreferredStockMember
2022-01-01
2022-12-31
0001560293
tnon:SeriesBConvertiblePreferredStockMember
us-gaap:PreferredStockMember
2022-01-01
2022-12-31
0001560293
us-gaap:CommonStockMember
2022-01-01
2022-12-31
0001560293
us-gaap:AdditionalPaidInCapitalMember
2022-01-01
2022-12-31
0001560293
us-gaap:RetainedEarningsMember
2022-01-01
2022-12-31
0001560293
us-gaap:AccumulatedOtherComprehensiveIncomeMember
2022-01-01
2022-12-31
0001560293
tnon:SeriesAConvertiblePreferredStockMember
us-gaap:PreferredStockMember
2022-12-31
0001560293
tnon:SeriesBConvertiblePreferredStockMember
us-gaap:PreferredStockMember
2022-12-31
0001560293
us-gaap:CommonStockMember
2022-12-31
0001560293
us-gaap:AdditionalPaidInCapitalMember
2022-12-31
0001560293
us-gaap:RetainedEarningsMember
2022-12-31
0001560293
us-gaap:AccumulatedOtherComprehensiveIncomeMember
2022-12-31
0001560293
tnon:SeriesAConvertiblePreferredStockMember
us-gaap:PreferredStockMember
2023-01-01
2023-12-31
0001560293
tnon:SeriesBConvertiblePreferredStockMember
us-gaap:PreferredStockMember
2023-01-01
2023-12-31
0001560293
us-gaap:CommonStockMember
2023-01-01
2023-12-31
0001560293
us-gaap:AdditionalPaidInCapitalMember
2023-01-01
2023-12-31
0001560293
us-gaap:RetainedEarningsMember
2023-01-01
2023-12-31
0001560293
us-gaap:AccumulatedOtherComprehensiveIncomeMember
2023-01-01
2023-12-31
0001560293
tnon:SeriesAConvertiblePreferredStockMember
us-gaap:PreferredStockMember
2023-12-31
0001560293
tnon:SeriesBConvertiblePreferredStockMember
us-gaap:PreferredStockMember
2023-12-31
0001560293
us-gaap:CommonStockMember
2023-12-31
0001560293
us-gaap:AdditionalPaidInCapitalMember
2023-12-31
0001560293
us-gaap:RetainedEarningsMember
2023-12-31
0001560293
us-gaap:AccumulatedOtherComprehensiveIncomeMember
2023-12-31
0001560293
tnon:SeriesAConvertiblePreferredStockMember
us-gaap:PreferredStockMember
2024-03-31
0001560293
us-gaap:CommonStockMember
2024-03-31
0001560293
us-gaap:AdditionalPaidInCapitalMember
2024-03-31
0001560293
us-gaap:RetainedEarningsMember
2024-03-31
0001560293
us-gaap:AccumulatedOtherComprehensiveIncomeMember
2024-03-31
0001560293
2024-03-31
0001560293
tnon:SeriesAConvertiblePreferredStockMember
us-gaap:PreferredStockMember
2024-04-01
2024-06-30
0001560293
us-gaap:CommonStockMember
2024-04-01
2024-06-30
0001560293
us-gaap:AdditionalPaidInCapitalMember
2024-04-01
2024-06-30
0001560293
us-gaap:RetainedEarningsMember
2024-04-01
2024-06-30
0001560293
us-gaap:AccumulatedOtherComprehensiveIncomeMember
2024-04-01
2024-06-30
0001560293
tnon:SeriesAConvertiblePreferredStockMember
us-gaap:PreferredStockMember
2024-06-30
0001560293
us-gaap:CommonStockMember
2024-06-30
0001560293
us-gaap:AdditionalPaidInCapitalMember
2024-06-30
0001560293
us-gaap:RetainedEarningsMember
2024-06-30
0001560293
us-gaap:AccumulatedOtherComprehensiveIncomeMember
2024-06-30
0001560293
tnon:SeriesAConvertiblePreferredStockMember
us-gaap:PreferredStockMember
2023-03-31
0001560293
us-gaap:CommonStockMember
2023-03-31
0001560293
us-gaap:AdditionalPaidInCapitalMember
2023-03-31
0001560293
us-gaap:RetainedEarningsMember
2023-03-31
0001560293
us-gaap:AccumulatedOtherComprehensiveIncomeMember
2023-03-31
0001560293
2023-03-31
0001560293
tnon:SeriesAConvertiblePreferredStockMember
us-gaap:PreferredStockMember
2023-04-01
2023-06-30
0001560293
us-gaap:CommonStockMember
2023-04-01
2023-06-30
0001560293
us-gaap:AdditionalPaidInCapitalMember
2023-04-01
2023-06-30
0001560293
us-gaap:RetainedEarningsMember
2023-04-01
2023-06-30
0001560293
us-gaap:AccumulatedOtherComprehensiveIncomeMember
2023-04-01
2023-06-30
0001560293
tnon:SeriesAConvertiblePreferredStockMember
us-gaap:PreferredStockMember
2023-06-30
0001560293
us-gaap:CommonStockMember
2023-06-30
0001560293
us-gaap:AdditionalPaidInCapitalMember
2023-06-30
0001560293
us-gaap:RetainedEarningsMember
2023-06-30
0001560293
us-gaap:AccumulatedOtherComprehensiveIncomeMember
2023-06-30
0001560293
2023-06-30
0001560293
tnon:SeriesAConvertiblePreferredStockMember
us-gaap:PreferredStockMember
2024-01-01
2024-06-30
0001560293
us-gaap:CommonStockMember
2024-01-01
2024-06-30
0001560293
us-gaap:AdditionalPaidInCapitalMember
2024-01-01
2024-06-30
0001560293
us-gaap:RetainedEarningsMember
2024-01-01
2024-06-30
0001560293
us-gaap:AccumulatedOtherComprehensiveIncomeMember
2024-01-01
2024-06-30
0001560293
tnon:SeriesAConvertiblePreferredStockMember
us-gaap:PreferredStockMember
2023-01-01
2023-06-30
0001560293
us-gaap:CommonStockMember
2023-01-01
2023-06-30
0001560293
us-gaap:AdditionalPaidInCapitalMember
2023-01-01
2023-06-30
0001560293
us-gaap:RetainedEarningsMember
2023-01-01
2023-06-30
0001560293
us-gaap:AccumulatedOtherComprehensiveIncomeMember
2023-01-01
2023-06-30
0001560293
2022-04-01
2022-04-06
0001560293
2021-05-01
2021-05-21
0001560293
2021-08-01
2021-08-31
0001560293
2021-10-01
2021-10-28
0001560293
2018-01-01
2018-12-31
0001560293
2019-01-01
2019-12-31
0001560293
2020-01-01
2020-12-31
0001560293
us-gaap:IPOMember
2023-01-01
2023-12-31
0001560293
us-gaap:RestrictedStockMember
2023-01-01
2023-12-31
0001560293
us-gaap:RestrictedStockMember
2022-01-01
2022-12-31
0001560293
us-gaap:EmployeeStockOptionMember
2023-01-01
2023-12-31
0001560293
us-gaap:EmployeeStockOptionMember
2022-01-01
2022-12-31
0001560293
us-gaap:WarrantMember
2023-01-01
2023-12-31
0001560293
us-gaap:WarrantMember
2022-01-01
2022-12-31
0001560293
us-gaap:FairValueInputsLevel2Member
us-gaap:CorporateDebtSecuritiesMember
2023-12-31
0001560293
us-gaap:FairValueInputsLevel2Member
us-gaap:CorporateDebtSecuritiesMember
2022-12-31
0001560293
us-gaap:CorporateDebtSecuritiesMember
2023-12-31
0001560293
us-gaap:CorporateDebtSecuritiesMember
2022-12-31
0001560293
us-gaap:ConstructionInProgressMember
2023-12-31
0001560293
us-gaap:ConstructionInProgressMember
2022-12-31
0001560293
tnon:CatamaranTraySetsMember
2023-12-31
0001560293
tnon:CatamaranTraySetsMember
2022-12-31
0001560293
us-gaap:EquipmentMember
2023-12-31
0001560293
us-gaap:EquipmentMember
2022-12-31
0001560293
us-gaap:LeaseholdImprovementsMember
2023-12-31
0001560293
us-gaap:LeaseholdImprovementsMember
2022-12-31
0001560293
tnon:LabEquipmentMember
2023-12-31
0001560293
tnon:LabEquipmentMember
2022-12-31
0001560293
us-gaap:OfficeEquipmentMember
2023-12-31
0001560293
us-gaap:OfficeEquipmentMember
2022-12-31
0001560293
tnon:ConvertibleNotePayableMember
2023-11-30
0001560293
us-gaap:WarrantMember
2023-11-30
0001560293
tnon:ConvertibleNotesMember
2023-12-31
0001560293
tnon:ConvertibleNotesMember
2023-01-01
2023-12-31
0001560293
us-gaap:ConvertibleDebtMember
us-gaap:SeriesAPreferredStockMember
2023-12-31
0001560293
tnon:ConvertibleNotesMember
us-gaap:SubsequentEventMember
2024-02-20
0001560293
us-gaap:WarrantMember
tnon:ConvertibleNotesMember
us-gaap:SubsequentEventMember
2024-02-20
0001560293
2014-02-18
0001560293
us-gaap:SeriesAPreferredStockMember
2023-01-01
2023-12-31
0001560293
us-gaap:SeriesBPreferredStockMember
2023-01-01
2023-12-31
0001560293
us-gaap:IPOMember
2022-04-26
2022-04-26
0001560293
us-gaap:IPOMember
2022-04-26
0001560293
tnon:UnderwritingAgreementMember
2022-04-26
0001560293
us-gaap:IPOMember
2022-04-29
2022-04-29
0001560293
tnon:ConvertibleNotePayableMember
2022-04-29
2022-04-29
0001560293
tnon:ConversionOfSeriesAAndSeriesBPreferredStockToCommonStockMember
2022-04-29
2022-04-29
0001560293
2021-05-31
2021-05-31
0001560293
tnon:AmendedAndRestatedExclusiveSalesRepresentativeAgreementMember
2021-05-31
2021-05-31
0001560293
us-gaap:IPOMember
2021-05-31
2021-05-31
0001560293
us-gaap:IPOMember
2021-05-31
0001560293
2023-06-16
0001560293
2023-06-16
2023-06-16
0001560293
tnon:OfferingWarrantsMember
2023-06-16
0001560293
2023-07-16
0001560293
2023-05-04
2023-05-04
0001560293
tnon:EquityDistributionAgreementMember
2023-01-01
2023-12-31
0001560293
tnon:LincolnParkCapitalFundLLCMember
2023-07-24
2023-07-24
0001560293
tnon:LincolnParkCapitalFundLLCMember
2023-01-01
2023-12-31
0001560293
tnon:LincolnParkCapitalFundLLCMember
2023-12-31
0001560293
tnon:EquityLineOfCreditMember
2023-12-31
0001560293
srt:MinimumMember
tnon:TwoThousandAndTwelvePlanMember
2023-01-01
2023-12-31
0001560293
srt:MaximumMember
tnon:TwoThousandAndTwelvePlanMember
2023-01-01
2023-12-31
0001560293
us-gaap:EmployeeStockOptionMember
tnon:TwoThousandAndTwelvePlanMember
2023-01-01
2023-12-31
0001560293
tnon:TwoThousandAndTwentyTwoEquityIncentivePlanMember
2022-01-10
2022-01-10
0001560293
tnon:TwoThousandAndTwentyTwoEquityIncentivePlanMember
2022-02-02
2022-02-02
0001560293
tnon:TwoThousandAndTwentyTwoEquityIncentivePlanMember
2023-01-01
2023-12-31
0001560293
us-gaap:WarrantMember
2022-04-30
0001560293
us-gaap:WarrantMember
2022-04-30
2022-04-30
0001560293
us-gaap:WarrantMember
us-gaap:AdditionalPaidInCapitalMember
2022-04-30
0001560293
us-gaap:WarrantMember
us-gaap:IPOMember
2022-04-30
0001560293
us-gaap:WarrantMember
2023-07-16
0001560293
us-gaap:WarrantMember
2023-07-14
2023-07-14
0001560293
us-gaap:WarrantMember
2023-01-01
2023-06-30
0001560293
us-gaap:WarrantMember
2023-11-30
2023-11-30
0001560293
us-gaap:WarrantMember
us-gaap:AdditionalPaidInCapitalMember
2023-11-30
0001560293
tnon:BlackScholesOptionMember
2023-01-01
2023-12-31
0001560293
tnon:BlackScholesOptionMember
2022-01-01
2022-12-31
0001560293
us-gaap:EmployeeStockOptionMember
2021-12-31
0001560293
us-gaap:EmployeeStockOptionMember
2021-12-31
2021-12-31
0001560293
us-gaap:RestrictedStockUnitsRSUMember
2021-12-31
0001560293
us-gaap:EmployeeStockOptionMember
2022-01-01
2022-12-31
0001560293
us-gaap:RestrictedStockUnitsRSUMember
2022-01-01
2022-12-31
0001560293
us-gaap:EmployeeStockOptionMember
2022-12-31
0001560293
us-gaap:RestrictedStockUnitsRSUMember
2022-12-31
0001560293
us-gaap:EmployeeStockOptionMember
2023-01-01
2023-12-31
0001560293
us-gaap:RestrictedStockUnitsRSUMember
2023-01-01
2023-12-31
0001560293
us-gaap:EmployeeStockOptionMember
2023-12-31
0001560293
us-gaap:RestrictedStockUnitsRSUMember
2023-12-31
0001560293
us-gaap:ResearchAndDevelopmentExpenseMember
2023-01-01
2023-12-31
0001560293
us-gaap:ResearchAndDevelopmentExpenseMember
2022-01-01
2022-12-31
0001560293
us-gaap:SellingAndMarketingExpenseMember
2023-01-01
2023-12-31
0001560293
us-gaap:SellingAndMarketingExpenseMember
2022-01-01
2022-12-31
0001560293
us-gaap:GeneralAndAdministrativeExpenseMember
2023-01-01
2023-12-31
0001560293
us-gaap:GeneralAndAdministrativeExpenseMember
2022-01-01
2022-12-31
0001560293
tnon:SalesRepresentativeAgreementMember
2020-04-01
2020-04-30
0001560293
tnon:SalesRepresentativeAgreementMember
2021-05-01
2021-05-31
0001560293
tnon:SalesRepresentativeAgreementMember
2021-05-31
0001560293
tnon:AmendedAndRestatedExclusiveSalesRepresentativeAgreementMember
2023-01-01
2023-12-31
0001560293
tnon:SalesRepresentativeAgreementMember
2021-10-31
0001560293
tnon:SalesRepresentativeAgreementMember
2022-04-30
0001560293
tnon:SalesRepresentativeAgreementMember
2023-12-31
0001560293
tnon:TerminationAgreementMember
2022-10-06
0001560293
tnon:TerminationAgreementMember
2022-10-06
2022-10-06
0001560293
country:US
tnon:TerminationAgreementMember
2022-10-06
0001560293
tnon:TerminationAgreementMember
2023-01-01
2023-12-31
0001560293
us-gaap:FairValueInputsLevel3Member
tnon:TerminationAgreementMember
2023-01-01
2023-12-31
0001560293
tnon:ConsultingAgreementMember
2022-10-06
2023-10-05
0001560293
tnon:ConsultingAgreementMember
2023-10-05
0001560293
currency:CHF
us-gaap:NetAssetsGeographicAreaMember
2023-12-31
0001560293
currency:CHF
us-gaap:NetAssetsGeographicAreaMember
2022-12-31
0001560293
us-gaap:CaliforniaFranchiseTaxBoardMember
2023-12-31
0001560293
us-gaap:WarrantMember
us-gaap:SeriesAPreferredStockMember
us-gaap:SubsequentEventMember
2024-02-20
0001560293
us-gaap:WarrantMember
us-gaap:CommonStockMember
us-gaap:SubsequentEventMember
2024-02-20
0001560293
us-gaap:SubsequentEventMember
2024-02-20
0001560293
us-gaap:SubsequentEventMember
2024-02-20
2024-02-20
0001560293
us-gaap:SeriesAPreferredStockMember
us-gaap:SubsequentEventMember
2024-02-20
0001560293
us-gaap:CommonStockMember
us-gaap:SubsequentEventMember
2024-02-20
0001560293
2024-02-20
0001560293
tnon:SeriesAConvertiblePreferredStockMember
2023-12-31
0001560293
us-gaap:ConvertiblePreferredStockMember
2023-12-31
0001560293
srt:MaximumMember
us-gaap:SeriesAPreferredStockMember
2023-01-01
2023-12-31
0001560293
srt:MinimumMember
us-gaap:SeriesAPreferredStockMember
2023-01-01
2023-12-31
0001560293
tnon:LiquidationMember
2023-12-31
0001560293
srt:ScenarioForecastMember
2024-03-29
0001560293
2024-05-06
0001560293
srt:MinimumMember
2023-12-21
2023-12-21
0001560293
srt:MaximumMember
2023-12-21
2023-12-21
0001560293
us-gaap:RestrictedStockMember
2024-01-01
2024-06-30
0001560293
us-gaap:RestrictedStockMember
2023-01-01
2023-06-30
0001560293
us-gaap:EmployeeStockOptionMember
2024-01-01
2024-06-30
0001560293
us-gaap:EmployeeStockOptionMember
2023-01-01
2023-06-30
0001560293
us-gaap:WarrantMember
2024-01-01
2024-06-30
0001560293
us-gaap:WarrantMember
2023-01-01
2023-06-30
0001560293
us-gaap:ConvertiblePreferredStockMember
2024-01-01
2024-06-30
0001560293
us-gaap:ConvertiblePreferredStockMember
2023-01-01
2023-06-30
0001560293
us-gaap:ConstructionInProgressMember
2024-06-30
0001560293
tnon:CatamaranTraySetsMember
2024-06-30
0001560293
us-gaap:EquipmentMember
2024-06-30
0001560293
us-gaap:LeaseholdImprovementsMember
2024-06-30
0001560293
tnon:LabEquipmentMember
2024-06-30
0001560293
us-gaap:OfficeEquipmentMember
2024-06-30
0001560293
tnon:ConvertibleNotePayableMember
2024-06-30
0001560293
tnon:ConvertibleNotePayableMember
2024-01-01
2024-06-30
0001560293
tnon:ConvertibleNotePayableMember
us-gaap:SeriesAPreferredStockMember
2024-06-30
0001560293
tnon:ConvertibleNotePayableMember
2024-02-20
0001560293
us-gaap:WarrantMember
tnon:ConvertibleNotePayableMember
2024-02-20
0001560293
us-gaap:SeriesAPreferredStockMember
2014-02-18
2014-02-18
0001560293
us-gaap:SeriesBPreferredStockMember
2014-02-18
2014-02-18
0001560293
us-gaap:CommonStockMember
tnon:AttheMarketOfferingProgramMember
2024-01-01
2024-06-30
0001560293
tnon:LincolnParkCapitalFundLLCMember
2024-01-01
2024-06-30
0001560293
tnon:LincolnParkCapitalFundLLCMember
2024-06-30
0001560293
tnon:EquityLineOfCreditMember
2024-01-01
2024-06-30
0001560293
tnon:PurchaseAgreementMember
tnon:SeriesAPreferredStockAndWarrantsMember
2024-02-20
2024-02-20
0001560293
tnon:PurchaseAgreementMember
tnon:SeriesAPreferredStockAndWarrantsMember
2024-02-20
0001560293
tnon:PurchaseAgreementMember
us-gaap:CommonStockMember
us-gaap:IPOMember
2024-02-20
0001560293
tnon:PurchaseAgreementMember
us-gaap:CommonStockMember
2024-02-20
2024-02-20
0001560293
tnon:ConvertibleNotePayableMember
2024-02-20
2024-02-20
0001560293
us-gaap:SeriesAPreferredStockMember
2024-01-01
2024-06-30
0001560293
tnon:SeriesAConvertiblePreferredStockMember
2024-06-30
0001560293
us-gaap:ConvertiblePreferredStockMember
2024-06-30
0001560293
srt:MaximumMember
us-gaap:SeriesAPreferredStockMember
2024-01-01
2024-06-30
0001560293
srt:MinimumMember
us-gaap:SeriesAPreferredStockMember
2024-01-01
2024-06-30
0001560293
tnon:MergerTransactionMember
2024-06-30
0001560293
tnon:TwoThousandTwelvePlanMember
us-gaap:CommonStockMember
2021-04-01
2021-04-30
0001560293
tnon:TwoThousandTwelvePlanMember
us-gaap:CommonStockMember
2021-07-01
2021-07-31
0001560293
srt:MinimumMember
tnon:TwoThousandTwelvePlanMember
us-gaap:CommonStockMember
2021-08-01
2021-08-31
0001560293
srt:MaximumMember
tnon:TwoThousandTwelvePlanMember
us-gaap:CommonStockMember
2021-08-01
2021-08-31
0001560293
tnon:TwoThousandAndTwentyTwoEquityIncentivePlanMember
us-gaap:CommonStockMember
2022-01-10
2022-01-10
0001560293
tnon:TwoThousandAndTwentyTwoEquityIncentivePlanMember
us-gaap:CommonStockMember
2022-02-02
2022-02-02
0001560293
2022-01-10
2022-01-10
0001560293
2022-02-02
2022-02-02
0001560293
us-gaap:RestrictedStockUnitsRSUMember
2024-04-08
2024-04-08
0001560293
2024-04-08
2024-04-08
0001560293
tnon:TwoThousandAndTwentyTwoEquityIncentivePlanMember
2024-01-01
2024-06-30
0001560293
tnon:UnderwritingAgreementMember
2022-04-30
0001560293
2023-06-01
2023-06-30
0001560293
us-gaap:WarrantMember
2023-07-16
2023-07-16
0001560293
us-gaap:WarrantMember
2023-06-01
2023-06-30
0001560293
tnon:PurchaseAgreementMember
tnon:SeriesAWarrantsMember
2024-02-20
0001560293
tnon:PurchaseAgreementMember
2024-02-20
2024-02-20
0001560293
tnon:PurchaseAgreementMember
us-gaap:WarrantMember
us-gaap:IPOMember
2024-02-20
0001560293
2024-04-08
0001560293
us-gaap:EmployeeStockOptionMember
2024-01-01
2024-06-30
0001560293
us-gaap:RestrictedStockUnitsRSUMember
2024-01-01
2024-06-30
0001560293
us-gaap:EmployeeStockOptionMember
2024-06-30
0001560293
us-gaap:RestrictedStockUnitsRSUMember
2024-06-30
0001560293
us-gaap:ResearchAndDevelopmentExpenseMember
2024-04-01
2024-06-30
0001560293
us-gaap:ResearchAndDevelopmentExpenseMember
2023-04-01
2023-06-30
0001560293
us-gaap:ResearchAndDevelopmentExpenseMember
2024-01-01
2024-06-30
0001560293
us-gaap:ResearchAndDevelopmentExpenseMember
2023-01-01
2023-06-30
0001560293
us-gaap:SellingAndMarketingExpenseMember
2024-04-01
2024-06-30
0001560293
us-gaap:SellingAndMarketingExpenseMember
2023-04-01
2023-06-30
0001560293
us-gaap:SellingAndMarketingExpenseMember
2024-01-01
2024-06-30
0001560293
us-gaap:SellingAndMarketingExpenseMember
2023-01-01
2023-06-30
0001560293
us-gaap:GeneralAndAdministrativeExpenseMember
2024-04-01
2024-06-30
0001560293
us-gaap:GeneralAndAdministrativeExpenseMember
2023-04-01
2023-06-30
0001560293
us-gaap:GeneralAndAdministrativeExpenseMember
2024-01-01
2024-06-30
0001560293
us-gaap:GeneralAndAdministrativeExpenseMember
2023-01-01
2023-06-30
0001560293
tnon:AmendedAndRestatedExclusiveSalesRepresentativeAgreementMember
2021-05-01
2021-05-31
0001560293
tnon:SalesRepresentativeAgreementMember
2024-06-30
0001560293
tnon:TerminationAgreementMember
2024-01-01
2024-06-30
0001560293
us-gaap:FairValueInputsLevel3Member
tnon:TerminationAgreementMember
2024-01-01
2024-06-30
iso4217:USD
iso4217:USD
xbrli:shares
xbrli:shares
xbrli:pure
We consent to the inclusion in this Form
S-1 Registration Statement of Tenon Medical, Inc. (the “Company”) of our report dated March 29, 2024, relating to our
audit of the Company’s consolidated financial statements as of December 31, 2023, and for the year then ended, included in the Company’s
Annual Report on Form 10-K for the fiscal year ended December 31, 2023, which report includes an explanatory paragraph expressing substantial
doubt regarding the Company’s ability to continue as a going concern.
